- Record: found
- Abstract: found
- Article: found
Metagenomics analysis revealed the distinctive ruminal microbiome and resistive profiles in dairy buffaloes

Read this article at
Abstract
Background
Antimicrobial resistance poses super challenges in both human health and livestock production. Rumen microbiota is a large reservoir of antibiotic resistance genes (ARGs), which show significant varations in different host species and lifestyles. To compare the microbiome and resistome between dairy cows and dairy buffaloes, the microbial composition, functions and harbored ARGs of rumen microbiota were explored between 16 dairy cows (3.93 ± 1.34 years old) and 15 dairy buffaloes (4.80 ± 3.49 years old) using metagenomics.
Results
Dairy buffaloes showed significantly different bacterial species (LDA > 3.5 & P < 0.01), enriched KEGG pathways and CAZymes encoded genes (FDR < 0.01 & Fold Change > 2) in the rumen compared with dairy cows. Distinct resistive profiles were identified between dairy cows and dairy buffaloes. Among the total 505 ARGs discovered in the resistome of dairy cows and dairy buffaloes, 18 ARGs conferring resistance to 16 antibiotic classes were uniquely detected in dairy buffaloes. Gene tcmA (resistance to tetracenomycin C) presented high prevalence and age effect in dairy buffaloes, and was also highly positively correlated with 93 co-expressed ARGs in the rumen ( R = 0.98 & P = 5E-11). In addition, 44 bacterial species under Lactobacillus genus were found to be associated with tcmA ( R > 0.95 & P < 0.001). L. amylovorus and L. acidophilus showed greatest potential of harboring tcmA based on co-occurrence analysis and tcmA-containing contigs taxonomic alignment.
Conclusions
The current study revealed distinctive microbiome and unique ARGs in dairy buffaloes compared to dairy cattle. Our results provide novel understanding on the microbiome and resistome of dairy buffaloes, the unique ARGs and associated bacteria will help develop strategies to prevent the transmission of ARGs.
Related collections
Most cited references72
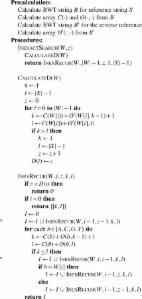
- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform

- Record: found
- Abstract: found
- Article: found
WGCNA: an R package for weighted correlation network analysis
- Record: found
- Abstract: found
- Article: found