- Record: found
- Abstract: found
- Article: found
Genomes of the cosmopolitan fruit pest Bactrocera dorsalis (Diptera: Tephritidae) reveal its global invasion history and thermal adaptation

Read this article at
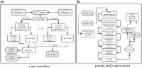
Graphical abstract

Highlights
-
•
The present study achieves a large-scale SNP dataset, a total of 512 genomes comprising 487 B. dorsalis and 25 B. carambolae.
-
•
B. dorsalis originates from the Southern India with three invasion routes worldwide, mainly facilitated by human activities.
-
•
CYP6a9 is identified that enhance the thermal adaptation of B. dorsalis and thus boost its invasion to temperate regions.
-
•
The gene function is further verified using the RNAi technology.
Abstract
Introduction
The oriental fruit fly Bactrocera dorsalis is one of the most destructive agricultural pests worldwide, with highly debated species delimitation, origin, and global spread routes.
Objectives
Our study intended to (i) resolve the taxonomic uncertainties between B. dorsalis and B. carambolae, (ii) reveal the population structure and global invasion routes of B. dorsalis across Asia, Africa, and Oceania, and (iii) identify genomic regions that are responsible for the thermal adaptation of B. dorsalis.
Methods
Based on a high-quality chromosome-level reference genome assembly, we explored the population relationship using a genome-scale single nucleotide polymorphism dataset generated from the resequencing data of 487 B. dorsalis genomes and 25 B. carambolae genomes . Genome-wide association studies and silencing using RNA interference were used to identify and verify the candidate genes associated with extreme thermal stress.
Results
We showed that B. dorsalis originates from the Southern India region with three independent invasion and spread routes worldwide: (i) from Northern India to Northern Southeast Asia, then to Southern Southeast Asia; (ii) from Northern India to Northern Southeast Asian, then to China and Hawaii; and (iii) from Southern India toward the African mainland, then to Madagascar, which is mainly facilitated by human activities including trade and immigration. Twenty-seven genes were identified by a genome-wide association study to be associated with 11 temperature bioclimatic variables. The Cyp6a9 gene may enhance the thermal adaptation of B. dorsalis and thus boost its invasion, which tended to be upregulated at a hardening temperature of 38 °C. Functional verification using RNA interference silencing against Cyp6a9, led to the specific decrease in Cyp6a9 expression, reducing the survival rate of dsRNA-feeding larvae exposed to extreme thermal stress of 45 °C after heat hardening treatments in B. dorsalis.
Related collections
Most cited references74

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools
- Record: found
- Abstract: found
- Article: not found
The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data.
- Record: found
- Abstract: found
- Article: found