- Record: found
- Abstract: found
- Article: not found
Tn-seq; high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms

Abstract
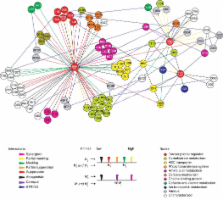
Biological pathways are structured in complex networks of interacting genes. Solving the architecture of such networks may provide valuable information, such as how microorganisms cause disease. Here we present a method (Tn-seq) for accurately determining quantitative genetic interactions on a genome-wide scale in microorganisms. Tn-seq is based on the assembly of a saturated Mariner transposon insertion library. After library selection, changes in frequency of each insertion mutant are determined by sequencing of the flanking regions en masse. These changes are used to calculate each mutant’s fitness. Fitness was determined for each gene of the gram-positive bacterium Streptococcus pneumoniae, a causative agent of pneumonia and meningitis. A genome-wide screen for genetic interactions identified both alleviating and aggravating interactions that could be further divided into seven distinct categories. Due to the wide activity of the Mariner transposon, Tn-seq has the potential to contribute to the exploration of complex pathways across many different species.
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles
- Record: found
- Abstract: found
- Article: not found
Genes required for mycobacterial growth defined by high density mutagenesis.
- Record: found
- Abstract: found
- Article: found