- Record: found
- Abstract: found
- Article: found
Open Babel: An open chemical toolbox
Read this article at
Abstract
Background
A frequent problem in computational modeling is the interconversion of chemical structures between different formats. While standard interchange formats exist (for example, Chemical Markup Language) and de facto standards have arisen (for example, SMILES format), the need to interconvert formats is a continuing problem due to the multitude of different application areas for chemistry data, differences in the data stored by different formats (0D versus 3D, for example), and competition between software along with a lack of vendor-neutral formats.
Results
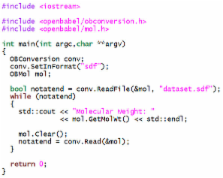
We discuss, for the first time, Open Babel, an open-source chemical toolbox that speaks the many languages of chemical data. Open Babel version 2.3 interconverts over 110 formats. The need to represent such a wide variety of chemical and molecular data requires a library that implements a wide range of cheminformatics algorithms, from partial charge assignment and aromaticity detection, to bond order perception and canonicalization. We detail the implementation of Open Babel, describe key advances in the 2.3 release, and outline a variety of uses both in terms of software products and scientific research, including applications far beyond simple format interconversion.
Conclusions
Open Babel presents a solution to the proliferation of multiple chemical file formats. In addition, it provides a variety of useful utilities from conformer searching and 2D depiction, to filtering, batch conversion, and substructure and similarity searching. For developers, it can be used as a programming library to handle chemical data in areas such as organic chemistry, drug design, materials science, and computational chemistry. It is freely available under an open-source license from http://openbabel.org.
Related collections
Most cited references66
- Record: found
- Abstract: found
- Article: not found
Very fast prediction and rationalization of pKa values for protein-ligand complexes.
- Record: found
- Abstract: not found
- Article: not found
Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions
- Record: found
- Abstract: not found
- Article: not found