- Record: found
- Abstract: found
- Article: found
Microbial metagenome-assembled genomes of the Fram Strait from short and long read sequencing platforms

Read this article at
Abstract
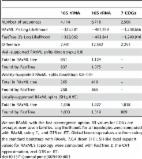
The impacts of climate change on the Arctic Ocean are manifesting throughout the ecosystem at an unprecedented rate. Of global importance are the impacts on heat and freshwater exchange between the Arctic and North Atlantic Oceans. An expanding Atlantic influence in the Arctic has accelerated sea-ice decline, weakened water column stability and supported the northward shift of temperate species. The only deep-water gateway connecting the Arctic and North Atlantic and thus, fundamental for these exchange processes is the Fram Strait. Previous research in this region is extensive, however, data on the ecology of microbial communities is limited, reflecting the wider bias towards temperate and tropical latitudes. Therefore, we present 14 metagenomes, 11 short-read from Illumina and three long-read from PacBio Sequel II, of the 0.2–3 µm fraction to help alleviate such biases and support future analyses on changing ecological patterns. Additionally, we provide 136 species-representative, manually refined metagenome-assembled genomes which can be used for comparative genomics analyses and addressing questions regarding functionality or distribution of taxa.
Related collections
Most cited references53
- Record: found
- Abstract: found
- Article: found
The SILVA ribosomal RNA gene database project: improved data processing and web-based tools
- Record: found
- Abstract: found
- Article: not found
MUSCLE: multiple sequence alignment with high accuracy and high throughput.
- Record: found
- Abstract: found
- Article: found