- Record: found
- Abstract: found
- Article: found
Realistic in silico generation and augmentation of single-cell RNA-seq data using generative adversarial networks
Read this article at
Abstract
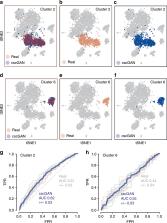
A fundamental problem in biomedical research is the low number of observations available, mostly due to a lack of available biosamples, prohibitive costs, or ethical reasons. Augmenting few real observations with generated in silico samples could lead to more robust analysis results and a higher reproducibility rate. Here, we propose the use of conditional single-cell generative adversarial neural networks (cscGAN) for the realistic generation of single-cell RNA-seq data. cscGAN learns non-linear gene–gene dependencies from complex, multiple cell type samples and uses this information to generate realistic cells of defined types. Augmenting sparse cell populations with cscGAN generated cells improves downstream analyses such as the detection of marker genes, the robustness and reliability of classifiers, the assessment of novel analysis algorithms, and might reduce the number of animal experiments and costs in consequence. cscGAN outperforms existing methods for single-cell RNA-seq data generation in quality and hold great promise for the realistic generation and augmentation of other biomedical data types.
Abstract
Low sample numbers often limit the robustness of analyses in biomedical research. Here, the authors introduce a method to generate realistic scRNA-seq data using GANs that learn gene expression dependencies from complex samples, and show that augmenting spare cell populations improves downstream analyses.
Related collections
Most cited references9
- Record: found
- Abstract: not found
- Conference Proceedings: not found
Image-to-Image Translation with Conditional Adversarial Networks

- Record: found
- Abstract: found
- Article: found
PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells
- Record: found
- Abstract: found
- Article: not found