- Record: found
- Abstract: found
- Article: found
Identification of six new susceptibility loci for invasive epithelial ovarian cancer
Karoline B Kuchenbaecker ,
Susan J Ramus ,
Jonathan Tyrer ,
Andrew Lee ,
Howard C Shen ,
Jonathan Beesley ,
Kate Lawrenson ,
Lesley McGuffog ,
Sue Healey ,
Janet M Lee ,
Tassja J Spindler ,
Yvonne G Lin ,
Tanja Pejovic ,
Yukie Bean ,
Qiyuan Li ,
Simon Coetzee ,
Dennis Hazelett ,
Alexander Miron ,
Melissa Southey ,
Mary Beth Terry ,
David E Goldgar ,
Saundra S Buys ,
Ramunas Janavicius ,
Cecilia M Dorfling ,
Elizabeth J van Rensburg ,
Susan L Neuhausen ,
Yuan Chun Ding ,
Thomas V O Hansen ,
Lars Jønson ,
Anne-Marie Gerdes ,
Bent Ejlertsen ,
Daniel Barrowdale ,
Joe Dennis ,
Javier Benitez ,
Ana Osorio ,
Maria Jose Garcia ,
Ian Komenaka ,
Jeffrey N Weitzel ,
Pamela Ganschow ,
Paolo Peterlongo ,
Loris Bernard ,
Alessandra Viel ,
Bernardo Bonanni ,
Bernard Peissel ,
Siranoush Manoukian ,
Paolo Radice ,
Laura Papi ,
Laura Ottini ,
Florentia Fostira ,
Irene Konstantopoulou ,
Judy Garber ,
Debra Frost ,
Jo Perkins ,
Radka Platte ,
Steve Ellis ,
Andrew K Godwin ,
Rita Katharina Schmutzler ,
Alfons Meindl ,
Christoph Engel ,
Christian Sutter ,
Olga M Sinilnikova ,
Francesca Damiola ,
Sylvie Mazoyer ,
Dominique Stoppa-Lyonnet ,
Kathleen Claes ,
Kim De Leeneer ,
Judy Kirk ,
Gustavo C Rodriguez ,
Marion Piedmonte ,
David M O'Malley ,
Miguel de la Hoya ,
Trinidad Caldes ,
Kristiina Aittomäki ,
Heli Nevanlinna ,
J Margriet Collée ,
Matti A Rookus ,
Jan C Oosterwijk ,
Laima Tihomirova ,
Nadine Tung ,
Ute Hamann ,
Claudine Isaccs ,
Marc Tischkowitz ,
Evgeny N Imyanitov ,
Maria A Caligo ,
Ian G Campbell ,
Frans B L Hogervorst ,
Edith Olah ,
Orland Diez ,
Ignacio Blanco ,
Joan Brunet ,
Conxi Lazaro ,
Miquel Angel Pujana ,
Anna Jakubowska ,
Jacek Gronwald ,
Jan Lubinski ,
Grzegorz Sukiennicki ,
Rosa B Barkardottir ,
Marie Plante ,
Jacques Simard ,
Penny Soucy ,
Marco Montagna ,
Silvia Tognazzo ,
Manuel R Teixeira ,
Vernon S Pankratz ,
Xianshu Wang ,
Noralane Lindor ,
Csilla I Szabo ,
Noah Kauff ,
Joseph Vijai ,
Carol A Aghajanian ,
Georg Pfeiler ,
Andreas Berger ,
Christian F Singer ,
Muy-Kheng Tea ,
Catherine M Phelan ,
Mark H Greene ,
Phuong L Mai ,
Gad Rennert ,
Anna Marie Mulligan ,
Sandrine Tchatchou ,
Irene L Andrulis ,
Gord Glendon ,
Amanda Ewart Toland ,
Uffe Birk Jensen ,
Torben A Kruse ,
Mads Thomassen ,
Anders Bojesen ,
Jamal Zidan ,
Eitan Friedman ,
Yael Laitman ,
Maria Soller ,
Annelie Liljegren ,
Brita Arver ,
Zakaria Einbeigi ,
Marie Stenmark-Askmalm ,
Olufunmilayo I Olopade ,
Robert L Nussbaum ,
Timothy R Rebbeck ,
Katherine L Nathanson ,
Susan M Domchek ,
Karen H Lu ,
Beth Y Karlan ,
Christine Walsh ,
Jenny Lester ,
Alexander Hein ,
Arif B Ekici ,
Matthias W Beckmann ,
Peter A Fasching ,
Diether Lambrechts ,
Els Van Nieuwenhuysen ,
Ignace Vergote ,
Sandrina Lambrechts ,
Ed Dicks ,
Jennifer A Doherty ,
Kristine G Wicklund ,
Mary Anne Rossing ,
Anja Rudolph ,
Jenny Chang-Claude ,
Shan Wang-Gohrke ,
Ursula Eilber ,
Kirsten B Moysich ,
Kunle Odunsi ,
Lara Sucheston ,
Shashi Lele ,
Lynne R Wilkens ,
Marc T Goodman ,
Pamela J Thompson ,
Yurii B Shvetsov ,
Ingo B Runnebaum ,
Matthias Dürst ,
Peter Hillemanns ,
Thilo Dörk ,
Natalia Antonenkova ,
Natalia Bogdanova ,
Arto Leminen ,
Liisa M Pelttari ,
Ralf Butzow ,
Francesmary Modugno ,
Joseph L Kelley ,
Robert P Edwards ,
Roberta B Ness ,
Andreas du Bois ,
Florian Heitz ,
Ira Schwaab ,
Philipp Harter ,
Keitaro Matsuo ,
Satoyo Hosono ,
Sandra Orsulic ,
Allan Jensen ,
Susanne Kruger Kjaer ,
Estrid Hogdall ,
Hanis Nazihah Hasmad ,
Mat Adenan Noor Azmi ,
Soo-Hwang Teo ,
Yin-Ling Woo ,
Brooke L Fridley ,
Ellen L Goode ,
Julie M Cunningham ,
Robert A Vierkant ,
Fiona Bruinsma ,
Graham G Giles ,
Dong Liang ,
Michelle A T Hildebrandt ,
Xifeng Wu ,
Douglas A Levine ,
Maria Bisogna ,
Andrew Berchuck ,
Edwin S Iversen ,
Joellen M Schildkraut ,
Patrick Concannon ,
Rachel Palmieri Weber ,
Daniel W Cramer ,
Kathryn L Terry ,
Elizabeth M Poole ,
Shelley S Tworoger ,
Elisa V Bandera ,
Irene Orlow ,
Sara H Olson ,
Camilla Krakstad ,
Helga B Salvesen ,
Ingvild L Tangen ,
Line Bjorge ,
Anne M van Altena ,
Katja K H Aben ,
Lambertus A Kiemeney ,
Leon F A G Massuger ,
Melissa Kellar ,
Angela Brooks-Wilson ,
Linda E Kelemen ,
Linda S Cook ,
Nhu D Le ,
Cezary Cybulski ,
Hannah Yang ,
Jolanta Lissowska ,
Louise A Brinton ,
Nicolas Wentzensen ,
Claus Hogdall ,
Lene Lundvall ,
Lotte Nedergaard ,
Helen Baker ,
Honglin Song ,
Diana Eccles ,
Ian McNeish ,
James Paul ,
Karen Carty ,
Nadeem Siddiqui ,
Rosalind Glasspool ,
Alice S Whittemore ,
Joseph H Rothstein ,
Valerie McGuire ,
Weiva Sieh ,
Bu-Tian Ji ,
Wei Zheng ,
Xiao-Ou Shu ,
Yu-Tang Gao ,
Barry Rosen ,
Harvey A Risch ,
John R McLaughlin ,
Steven A Narod ,
Alvaro N Monteiro ,
Ann Chen ,
Hui-Yi Lin ,
Jenny Permuth-Wey ,
Thomas A Sellers ,
Ya-Yu Tsai ,
Zhihua Chen ,
Argyrios Ziogas ,
Hoda Anton-Culver ,
Aleksandra Gentry-Maharaj ,
Usha Menon ,
Patricia Harrington ,
Alice W Lee ,
Anna H Wu ,
Celeste L Pearce ,
Gerry Coetzee ,
Malcolm C Pike ,
Agnieszka Dansonka-Mieszkowska ,
Agnieszka Timorek ,
Iwona K Rzepecka ,
Jolanta Kupryjanczyk ,
Matt Freedman ,
Houtan Noushmehr ,
Douglas F Easton ,
Kenneth Offit ,
Fergus J Couch ,
Simon Gayther ,
Paul P Pharoah ,
Antonis C Antoniou ,
Georgia Chenevix-Trench ,
EMBRACE,
GEMO Study Collaborators,
Breast Cancer Family Registry,
HEBON,
KConFab Investigators,
Australian Cancer Study (Ovarian Cancer Investigators),
Australian Ovarian Cancer Study Group,
the Consortium of Investigators of Modifiers of BRCA1 and BRCA2
January 12 2015
Read this article at
ScienceOpenPublisher
- oa repository (via OAI-PMH doi match)
- oa repository (via OAI-PMH doi match)
- oa repository (via OAI-PMH title and first author match)
- oa repository (via pmcid lookup)
- oa repository (via OAI-PMH doi match)
- oa repository (via OAI-PMH doi match)
- oa repository (via OAI-PMH title and first author match)
Powered by 
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Genome-wide association studies (GWAS) have identified 12 epithelial ovarian cancer
(EOC) susceptibility alleles. The pattern of association at these loci is consistent
in BRCA1 and BRCA2 mutation carriers who are at high EOC risk. After imputation to
the 1000 Genomes Project data, we assessed associations of 11 million genetic variants
with EOC risk from 15,397 cases unselected for family history and 30,816 controls,
15,252 BRCA1 mutation carriers and 8,211 BRCA2 mutation carriers (3,096 with ovarian
cancer), and combined the results in a meta-analysis. This new study design yielded
increased statistical power, leading to the discovery of six new EOC susceptibility
loci. Variants at 1p36 (nearest gene WNT4), 4q26 (SYNPO2), 9q34.2 (ABO) and 17q11.2
(ATAD5) were associated with EOC risk, and at 1p34.3 (RSPO1) and 6p22.1 (GPX6) specifically
with the serous EOC subtype, at p<5×10−8. Incorporating these variants into risk assessment
tools will improve clinical risk predictions for BRCA1/2 mutation carriers.
The risk of developing invasive EOC is higher than the population average for relatives
of women diagnosed with the disease
1,2
, indicating the importance of genetic factors in disease susceptibility. Approximately
25% of the familial aggregation of EOC is explained by rare, high-penetrance alleles
of BRCA1 and BRCA2
3
. Furthermore, population-based GWAS have identified common variants associated with
invasive EOC at 11 loci
4–9
but only six have also been evaluated in BRCA1 and/or BRCA2 mutation carriers. All
loci displayed associations in mutation carriers that were consistent with the associations
observed in the general population
10–12
. In addition, the 4q32.3 locus is associated with EOC risk for BRCA1 mutation carriers
only
13
. However, the common genetic variants explain less than 3.1% of the excess familial
risk of EOC so additional susceptibility loci are likely to exist.
Women diagnosed with EOC and unaffected women from the general population ascertained
through the Ovarian Cancer Association Consortium (OCAC)
14
and BRCA1 and BRCA2 mutation carriers from the Consortium of Investigators of Modifiers
of BRCA1/2 (CIMBA)
15
were genotyped as part of the Collaborative Oncological Gene-environment Study (COGS)
using the iCOGS custom array. In addition, data were available for cases and controls
from three EOC GWAS. We first evaluated whether the EOC susceptibility loci at 8q21.13,
10p12.31, 17q12, 5p15.33, and 17q21.31 recently identified by OCAC
7–9
also show evidence of association in BRCA1 and BRCA2 mutation carriers. Using data
from >200,000 genotyped SNPs
7,13,16
, we performed imputation of common variants from the 1000 Genomes Project data
17
and evaluated the associations of these SNPs with invasive EOC risk in OCAC and in
BRCA1 and BRCA2 mutation carriers from CIMBA. Given the strong evidence for a significant
overlap in loci predisposing to EOC in the general population and those associated
with risk in BRCA1 and BRCA2 mutation carriers, we carried out a meta-analysis of
the EOC risk associations in order to identify novel EOC susceptibility loci.
Genotype data were available for imputation on 15,252 BRCA1 mutation carriers and
8,211 BRCA2 mutation carriers, of whom 2,462 and 631, respectively, were affected
with EOC
13,16
. From OCAC, genotyping data were available from 15,437 women with invasive EOC (including
9,627 with serous EOC) and 30,845 controls from the general population
7
. Imputation was performed separately for BRCA1 carriers, BRCA2 carriers, OCAC-COGS
samples and the three OCAC GWAS (Supplementary Tables 1–2; Supplementary Fig. 1; Supplementary
Fig. 2). The meta-analysis was based on 11,403,952 SNPs (Supplementary Fig. 3).
Of five EOC susceptibility loci that have not yet been evaluated in mutation carriers,
two were associated with EOC risk for both BRCA1 and BRCA2 mutation carriers at p<0.05
(10p12.31 and 17q21.31) (Supplementary Table 3). Overall, seven of the twelve known
EOC susceptibility loci provided evidence of association in BRCA1 mutation carriers
and six were associated in BRCA2 mutation carriers. However, with the exception of
5p15.33 (TERT), all loci had hazard ratio (HR) estimates in BRCA1 and BRCA2 carriers
that were in the same direction as the odds ratio (OR) estimates for serous subtype
EOC from OCAC (Fig. 1). Analysing the associations jointly in BRCA1 and BRCA2 carriers
and serous EOC in OCAC provided stronger evidence of association, with smaller p-values
for eight of the susceptibility variants compared to the analysis in OCAC alone.
Using the imputed genotypes, we observed no novel associations at p<5×10−8 in the
analysis of associations in BRCA1 or BRCA2 mutation carriers separately. However,
we identified seven previously unreported associations (p-values<5×10−8) in either
OCAC alone, the meta-analysis of EOC associations in BRCA1, BRCA2 carriers and OCAC,
or in the meta-analysis in BRCA1 and BRCA2 carriers and serous EOC in OCAC (Supplementary
Fig. 4; Supplementary Tables 4–5). SNPs in six of these loci remained genome-wide
statistically significant after re-imputing genotypes with imputation parameters set
to maximise accuracy (Table 1; Fig. 1). SNPs at 17q11.2 (near ATAD5) were found to
be associated with invasive EOC in OCAC (p<5×10−8) (Table 1). For the lead SNP, chr17:29181220:I,
the estimated HR estimate for BRCA1 mutation carriers was significantly different
from the estimate in OCAC (p=0.005); the association for BRCA2 carriers was consistent
with the OCAC OR estimate (BRCA2-OCAC meta-analysis p=2.6×10−9). SNPs at four loci
were associated at p<5×10−8 with risk of all invasive EOC in the meta-analysis (Supplementary
Fig. 5): 1p36, 1p34.3, 4q26, and 9q34.2. At 1p34.3, the most strongly associated SNP,
rs58722170, displayed stronger associations in the meta-analysis of serous EOC for
OCAC (p=2.7×10−12). In addition, SNPs at 6p22.1 were associated at genome-wide significance
level in the meta-analysis of associations with serous EOC (p=3.0×10−8), but not in
the meta-analysis of all invasive EOC associations (p=6.8×10−6).
The most significantly associated SNP at each of the six novel loci had high imputation
accuracy (r2≥0.83). At the 1p34.3, 1p36, and 6p22.1 loci, there was at least one genome-wide
significant genotyped SNP correlated with the lead SNP (pairwise r2≥0.73) (Supplementary
Table 6; Supplementary Fig. 5; Supplementary Note). We genotyped the leading (imputed)
SNPs of the three other loci in a subset of the samples using iPLEX (Supplementary
Note). The correlations between the expected allele dosages from the imputation and
the observed genotypes for the variants at 4q26 and 9q34.2, (r2=0.90 and r2=0.84,
respectively) were consistent with the estimated imputation accuracy (0.93 and 0.83
for CIMBA samples). The lead SNP at 17q11.2 failed iPLEX design. However, the risk
allele is highly correlated with the AA haplotype of two genotyped variants on the
iCOGS array (rs9910051 and rs3764419). This haplotype is strongly associated with
ovarian cancer risk in the subset of samples genotyped using iCOGS (BRCA2-OCAC meta-analysis
p=8.6×10−8 for haplotype, and p=1.8×10−8 for chr17:29181220:I) (Supplementary Table
7).
None of the regions contained additional SNPs that displayed EOC associations at p<10−4
in OCAC, BRCA1 carriers or BRCA2 carriers in multi-variable analyses adjusted for
the lead SNP in each region, indicating that they each contain only one independent
set of correlated highly associated variants (iCHAV). Relative to the 1000 Genomes
Project data, we had genotyped or imputed data covering 91% of the genetic variation
at 1p36, 84% at 1p34.3 and 83% at 4q26. The other three novel loci had coverage of
less than 80% (Supplementary Note). There was evidence for heterogeneity at p<0.05
in the associations with histological subtype in OCAC for the lead SNPs at 1p34.4
and 6p22.1, but not for at 1p36, 4q26, 9q34.2 and 17q11.2 (Table 2).
We carried out a competing risks association analysis in BRCA1 and BRCA2 mutation
carriers in order to investigate whether these loci are also associated with breast
cancer risk for mutation carriers (Supplementary Note). We used the most strongly
associated genotyped SNPs for this purpose because the statistical method requires
actual genotypes
18
. The EOC HR estimates were consistent with the estimates from the main analysis for
all SNPs (Supplementary Table 8). None of the SNPs displayed associations with breast
cancer risk at p<0.05.
At each of the six loci, we identified a set of SNPs with odds of less than 100 to
1 against being the causal variant; most are in non-coding DNA regions (Supplementary
Table 9). None were predicted to have likely deleterious functional effects although
some lie in or near chromatin biofeatures in fallopian tube and ovarian epithelial
cells which may represent the functional regulatory targets of the risk SNPs (Table
3; Supplementary Table 10). We also evaluated the protein coding genes in each region
for their role in EOC development, and as candidate susceptibility gene targets. Molecular
profiling data from 496 HGSOCs performed by The Cancer Genome Atlas (TCGA) indicated
frequent loss/deletion at four risk loci (1p36, 4q26, 9q34.2 and 17q11.2) (Supplementary
Table 11). Consistent with this, WNT4 and ABO were significantly down-regulated in
ovarian tumours while ATAD5 was up-regulated. Somatic coding sequence mutations in
the six genes nearest the index SNPs were rare. We performed expression quantitative
trait locus (eQTL) analysis in a series of 59 normal ovarian tissues (Supplementary
Table 12) to evaluate the gene nearest the top ranked SNP at each locus. For the five
genes expressed in normal cells, we found no statistically significant eQTL associations
for any of the putative causal SNPs at each locus; neither did we find any significant
tumour-eQTL associations for these genes based on data from TCGA (Supplementary Table
12). At the 1p36 locus, the most strongly associated variant, rs56318008, is located
in the promoter region of WNT4 which encodes a ligand in the WNT signal transduction
pathway, critical for cell proliferation and differentiation. Using a luciferase reporter
assay we found no effect of these putatively causal SNPs on WNT4 transcription in
iOSE4 normal ovarian cells (Fig. 2). Some of the putative causal SNPs at 1p36 are
located in CDC42 and LINC00339, and several are in putative regulatory domains in
ovarian tissues (Supplementary Table 10; Fig. 2). CDC42 is known to play a role in
migration and signalling in ovarian and breast cancer
19,20
. SNPs at 1p36 are also associated with increased risk of endometriosis and WNT4,
CDC42 and LINC00339 have all been implicated in endometriosis
21
, a known risk factor for endometrioid and clear cell EOC
22
.
The strongest associated variant at 1q34, rs58722170, is located in RSPO1, which encodes
R-spondin 1, a protein involved in cell proliferation (Supplementary Fig. 6). RSPO1
is important in tumorigenesis and early ovarian development
23,24
, and regulates WNT4 expression in the ovaries
25
. SYNPO2 at 4q26 encodes myopodin which is involved in cell motility and growth
26
and has a reported tumour suppressor role
27–30
. rs635634 is located upstream of the ABO gene (Supplementary Fig. 7). A moderately
correlated variant (rs505922, r2=0.52) determines ABO blood group and is associated
with increased risk of pancreatic cancer
31,32
. Previous studies in OCAC also showed a modestly increased risk of EOC for individuals
with the A blood group
33
. The moderate correlation between rs635634 and rs505922 and considerably weaker EOC
association of rs505922 (p=1.2×10−5) suggests that the association with blood group
is probably not driving the association with risk. The indel, 17:29181220:I, at 17q11.2
is located in ATAD5 which acts as a tumour suppressor gene
34–36
(Supplementary Fig. 8). ATAD5 modulates the interaction between RAD9A and BCL2 in
order to induce DNA damage related apoptosis. Finally, rs116133110, at 6p22.1, lies
in GPX6 which has no known role in cancer.
The six novel loci reported in this study increase the number of genome-wide significant
common variant loci so far identified for EOC to 18. Taken together, these explain
approximately 3.9% of the excess familial relative risk of EOC in the general population,
and account for approximately 5.2% of the EOC polygenic modifying variance in BRCA1
mutation carriers and 9.3% in BRCA2 mutation carriers. The similarity in the magnitude
of associations between BRCA1 and BRCA2 carriers and population-based studies suggests
a general model of susceptibility whereby BRCA1 and BRCA2 mutations and common alleles
interact multiplicatively on the relative risk scale for EOC
37
. This model predicts large differences in absolute EOC risk between individuals carrying
many alleles and individuals carrying few risk alleles of EOC susceptibility loci
for BRCA1 and BRCA2 mutation carriers
13,16
. Incorporating EOC susceptibility variants into risk assessment tools will improve
risk prediction and may be particularly useful for BRCA1 and BRCA2 mutation carriers.
METHODS
Study populations
We obtained data on BRCA1 and BRCA2 mutation carriers through CIMBA. Eligibility in
CIMBA is restricted to females 18 years or older with pathogenic mutations in BRCA1
or BRCA2. The majority of the participants were sampled through cancer genetics clinics
15
, including some related participants. Fifty-four studies from 27 countries contributed
data. After quality control, data were available on 15,252 BRCA1 mutation carriers
and 8,211 BRCA2 mutation carriers, of whom 2,462 and 631, respectively, were affected
with EOC (Supplementary Table 1).
Data were available for the stage 1 of three population-based EOC GWAS. These included
2,165 cases and 2,564 controls from a GWAS from North America (“US GWAS”)
39
, 1,762 cases and 6,118 controls from a UK-based GWAS (“UK GWAS”)
6
, and 441 cases and 441 controls from the Mayo GWAS. Furthermore, 11,069 cases and
21,722 controls were genotyped using the iCOGS array (“OCAC-iCOGS” stage data). Overall,
43 studies from 11 countries provided data on 15,347 women diagnosed with invasive
epithelial EOC, 9,627 of whom were diagnosed with serous EOC, and 30,845 controls
from the general population.
All subjects included in this analysis were of European descent and provided written
informed consent as well as data and blood samples under ethically approved protocols.
Further details of the OCAC and CIMBA study populations as well as the genotyping,
quality control and statistical analyses have been described elsewhere
7,13,16
.
Genotype data
Genotyping and imputation details for each study are shown in Supplementary Table
1.
Confirmatory genotyping of imputed SNPs
To evaluate the accuracy of the imputation of the SNPs we found to be associated with
EOC risk, we genotyped rs17329882 (4q26) and rs635634 (9q34.2) in a subset of 3,541
subjects from CIMBA using Sequenon’s iPLEX technology. The lead SNP at 17q11.2, chr17:29181220:I
failed iPLEX design. We performed quality control of the iPLEX data according to the
CIMBA guidelines. After quality control, we used the imputation results to generate
the expected allele dosage for each genotyped sample and computed the Pearson product-moment
correlation coefficient between the expected allele dosage and the observed genotype.
The squared correlation coefficient was compared to the imputation accuracy as estimated
from the imputation.
Quality control of GWAS and iCOGS genotyping data
We carried out quality control separately for BRCA1 carriers, BRCA2 carriers, the
three OCAC GWAS, and OCAC-iCOGS samples, but quality criteria were mostly consistent
across studies. We excluded samples if they were not of European ancestry, if they
had a genotyping call rate < 95%, low or high heterozygosity, if they were not female
or had ambiguous sex, or were duplicates (cryptic or intended). In OCAC studies, one
individual was excluded from each pair of samples found to be first-degree relatives
and duplicate samples between the iCOGS stage and any of the GWAS were excluded from
the iCOGS data. SNPs were excluded if they were monomorphic, had call rate<95%, showed
evidence of deviation from Hardy-Weinberg equilibrium or had low concordance between
duplicate pairs. For the Mayo GWAS and the UK GWAS, we also excluded rare SNPs (MAF<1%
or allele count <5, respectively). We visually inspected genotype cluster plots for
all SNPs with P<10−5 from each of the newly identified loci. We used the R GenABEL
library version 1.6.7 for quality control
40
.
Genotype data were available for analysis from iCOGS for 199,526 SNPs in OCAC-iCOGS,
200,720 SNPs in BRCA1 mutation carriers, and 200,908 SNPs in BRCA2 mutation carriers.
After QC, for the GWAS, data were available on 492,956 SNPs for the US GWAS, 543,529
SNPs for the UK GWAS and 1,587,051 SNPs for the Mayo GWAS (Supplementary Table 2).
Imputation
We performed imputation separately for BRCA1 carriers, BRCA2 carriers, OCAC-iCOGS
samples and each of the OCAC GWAS. We imputed variants from the 1000 Genomes Project
data using the v3 April 2012 release
17
as the reference panel. For OCAC-iCOGS, the UK GWAS and the Mayo GWAS, imputation
was based on the 1000 Genomes Project data with singleton sites removed. To improve
computation efficiency we initially used a two-step procedure, which involved pre-phasing
in the first step and imputation of the phased data in the second. We carried out
pre-phasing using the SHAPEIT software
41
. We used the IMPUTE version 2 software for the subsequent imputation
42
for all studies with the exception of the US GWAS for which the MACH algorithm implemented
in the minimac software version 2012.8.15, mach version 1.0.18 was used. To perform
the imputation we divided the data into segments of approximately 5Mb each. We excluded
SNPs from the association analysis if their imputation accuracy was r2<0.3 or their
minor allele frequency (MAF) was <0.005 in BRCA1 or BRCA2 carriers or if their accuracy
was r2<0.25 in OCAC-iCOGS, the UK GWAS, UK GWAS or Mayo GWAS.
We performed more accurate imputation for the regions around the novel EOC loci from
the joint analysis of the data from BRCA1 and BRCA2 carriers and the general population
(any SNP with P<5×10−8). The boundaries of these regions were set +/− 500kb from any
significantly associated SNP in the region. As in the first run, the 1000 Genomes
Project data v3 were used as the reference panel and the software IMPUTE2 was applied.
However, for the second round of imputation, we imputed genotypes without pre-phasing
in order to improve accuracy. To further increase the imputation accuracy we changed
some of the default parameters in the imputation procedure. These included an increase
of the MCMC iterations to 90 (out of which the first 15 were used as burn-in), an
increase of the buffer region to 500kb and an increase of the number of haplotypes
used as templates when phasing observed genotypes to 100. These changes were applied
consistently for all data sets.
Statistical analyses
Association analyses in the unselected ovarian cancer cases and controls from OCAC
We evaluated the association between genotype and disease using logistic regression
by estimating the associations with each additional copy of the minor allele (log-additive
models). The analysis was adjusted for study and for population substructure by including
the eigenvectors of the first five ancestry specific principal components as covariates
in the model. We used the same approach to evaluate the SNP associations with serous
ovarian cancer after excluding all cases with any other or with unknown tumour subtype.
For imputed SNPs we used expected dosages in the logistic regression model to estimate
SNP effect sizes and p-values. We carried out analyses separately for OCAC-iCOGS and
the three GWAS and pooled thereafter using a fixed effects meta-analysis. We carried
out the analysis of re-imputed genotypes of putative novel susceptibility loci jointly
for the OCAC-iCOGS and GWAS samples. All results are based on the combined data from
iCOGS and the three GWAS. We used custom written software for the analysis.
Associations in BRCA1 and BRCA2 mutation carriers from CIMBA
We carried out the ovarian cancer association analyses separately for BRCA1 and BRCA2
mutation carriers. The primary analysis was carried out within a survival analysis
framework with time to ovarian cancer diagnosis as the endpoint. Mutation carriers
were followed until the age of ovarian cancer diagnosis, or risk-reducing salpingo-oophorectomy
(RRSO) or age at last observation. Breast cancer diagnosis was not considered as a
censoring event. In order to account for the non-random sampling of BRCA1 and BRCA2
mutation carriers with respect to their disease status we conducted the analyses by
modelling the retrospective likelihood of the observed genotypes conditional on the
disease phenotype
18
. We assessed the associations between genotype and risk of ovarian cancer using the
1 degree of freedom score test statistic based on the retrospective likelihood
18,43
. To account for the non-independence among related individuals in the sample, we
used an adjusted version of the score test statistic, which uses a kinship adjusted
variance of the score
44
. We evaluated associations between imputed genotypes and ovarian cancer risk using
a version of the score test as described above but with the posterior genotype probabilities
replacing the genotypes. All analyses were stratified by the country of origin of
the samples.
We carried out the retrospective likelihood analyses in CIMBA using custom written
functions in Fortran and Python. The score test statistic was implemented in R version
3.0.1
45
.
We evaluated whether there is evidence for multiple independent association signals
in the region around each newly identified locus by evaluating the associations of
genetic variants in the region while adjusting for the SNP with the smallest meta-analysis
p-value in the respective region. This was done separately for BRCA1 carriers, BRCA2
carriers and OCAC.
For one of the novel associations, it was not possible to confirm the imputation accuracy
of the lead SNP chr17:29181220:I at 17q11.2 through genotyping. Therefore, we inferred
two-allele haplotypes for rs9910051 and rs3764419, highly correlated with the lead
SNP (r2=0.95), using an in-house program. These variants were genotyped on the iCOGS
array and therefore this analysis was restricted to 14,733 ovarian cancer cases and
9,165 controls from OCAC-COGS, and 8,185 BRCA2 mutation carriers that had available
genotypes for both variants based on iCOGS. The association between the AA haplotype
and risk was tested using logistic regression in OCAC and using Cox regression in
BRCA2 mutation carriers.
Meta-analysis
We conducted a meta-analysis of the EOC associations in BRCA1, BRCA2 carriers and
the general population for genotyped and imputed SNPs using an inverse variance approach
assuming fixed effects. We combined the logarithm of the per-allele hazard ratio estimate
for the association with EOC risk in BRCA1 and BRCA2 mutation carriers and the logarithm
of the per-allele odds ratio estimate for the association with disease status in OCAC.
For the associations in BRCA1 and BRCA2 carriers, we used the kinship adjusted variance
estimator
44
which allows for inclusion of related individuals in the analysis. We only used SNPs
with results in OCAC and in at least one of the BRCA1 or the BRCA2 analyses. We carried
out two separate meta-analyses, one for the associations with EOC in BRCA1 carriers,
BRCA2 carriers and EOC in OCAC, irrespective of tumour histological subtype, and a
second using only the associations with serous EOC in OCAC. The number of BRCA1 and
BRCA2 samples with tumour histology information was too small to allow for subgroup
analyses. However, previous studies have demonstrated that the majority of EOCs in
BRCA1 and BRCA2 mutation carriers are high-grade serous
49–53
. Meta-analyses were carried out using the software “metal”, 2011-03-25 release
54
.
Candidate causal SNPs in each susceptibility region
In order to identify a set of potentially causal variants we excluded SNPs with a
likelihood of being causal of less than 1:100, by comparing the likelihood of each
SNP from the association analysis with the one of the most strongly associated SNP
46
. The remaining variants were then analysed using pupasuite 3.1 to identify potentially
functional variants (Supplementary Table 9).
Functional analysis
Expression quantitative trait locus (eQTL) analysis in normal OSE and FTSE cells
Early-passage primary normal ovarian surface epithelial cells (OSECs) and fallopian
tube epithelial cells were harvested from disease-free ovaries and fallopian tubes.
Normal ovarian epithelial cells were collected by brushing the surface of the ovary
with a sterile cytobrush, and were cultured in NOSE-CM
55
. Fallopian tube epithelial cells were harvested by Pronase digestion as previously
described
56
, plated onto collagen-coated plastics (Sigma) and cultured in DMEM/F12 (Sigma-Aldrich)
supplemented with 2% Ultroser G (BioSepra) and 1× penicillin/streptomycin (Lonza).
By the time of RNA harvesting, fallopian tube cultures tested consisted of PAX8 positive
fallopian tube secretory epithelial cells (FTSECs), consistent with previous observations
that ciliated epithelial cells from the fallopian tube do not proliferate in vitro.
For gene expression analysis, RNA was harvested from 59 early passage samples: 54
OSECs and 5 FTSECs from cell cultures harvested at ~80% confluency using the QIAgen
miRNAeasy kit with on-column DNase 1 digestion. 500ng RNA was reverse transcribed
using the Superscript III kit (Life Technologies). We preamplified 10ng cDNA using
the TaqMan® Preamp Mastermix; the resulting product was diluted 1:60 and used to quantify
gene expression using the following TaqMan® gene expression probes: WNT4, Hs01573504_m1;
RSPO1, Hs00543475_m1; SYNPO2, Hs00326493_m1; ATAD5, Hs00227495_m1 and GPX6, Hs00699698_m1.
Four control genes were also included: ACTB, Hs00357333_g1; GAPDH, Hs02758991_g1;
HMBS, Hs00609293_g1 and HPRT1 Hs02800695_m1 (all Life Technologies). Assays were run
on an ABI 7900HT Fast Real-Time PCR system (Life Technologies).
Data Analysis
Expression levels for each gene were normalized to the average of all four control
genes. Relative expression levels were calculated using the δδCt method. Genotyping
was performed on the iCOGs chips, as described above. Where genotyping data were not
available for the most risk-associated SNP, the next most significant SNP was used:
rs3820282 at 1p36, rs12023270 at 1p34.3, rs752097 at 4q26, rs445870 at 6p22.1, rs505922
at 9q34.2 and rs3764419 at 17q11.2. Correlations between genotype and gene expression
were calculated in ‘R’. Genotype specific gene expression in the normal tissue cell
lines (eQTL analysis) was compared using the Jonckheere-Terpstra test. IData were
normalized to the four control genes and we tested for eQTL associations, grouping
OSECs and FTSECs together. Secondly, OSECs were analysed alone. eQTL analyses were
performed using 3 genotype groups, or two groups (with the rare homozygote samples
grouped together with the heterozygote samples).
eQTL analysis in primary ovarian tumours
eQTL analysis in primary tumours was based on the publicly available data available
from The Cancer Genome Atlas (TCGA) project, which includes 489 primary high grade
serous ovarian cancers. The methods have been described elsewhere
57
. Briefly, we determined the ancestry for each case based on the germ line genotype
data using EIGENSTRAT software with 415 HapMap genotype profiles as a control set.
Only populations of Northern and Western European ancestries were included. We first
performed a cis-eQTL analyses using a method we described previously, in which the
association between 906,600 germline genotypes and the expression levels of mRNA or
miRNA (located within 500Kb on either side of the variant) were evaluated using linear
regression model with the effects of somatic copy number and CpG methylation being
deducted (For miRNA expression, the effect of CpG methylation is not adjusted for
since the data are not available). To adjust for multiple tests, we adjusted the test
P values using Benjamini-Hochberg method. A significant association was defined by
a false discovery rate (FDR) of less than 0.1.
Having established a genome-wide cis-eQTL associaitions in this series of tumours,
we then evaluated cis-eQTL associations for the top risk associations between each
of the six new loci and the gene in closest proximity to the risk SNP. For each risk
locus, we retrieved the genotype of all SNPs in ovarian cancer cases based on the
Affymetrix 6.0 array. Using these genotypes and the impute2 March 2012 1000 Genomes
Phase I integrated variant cosmopolitan reference panel of 1,092 individuals (Haplotypes
were phased via SHAPEIT), we imputed the genotypes of SNPs in the 1000 Genomes Project
in the target regions for TCGA samples
58
. For each risk locus where data for the most risk-associated variant were not available,
we retrieved the imputed variants tightly correlated with the most risk-associated
variant. We then tested for association between imputed SNPs and gene expression using
the linear regression algorithm described above, where each imputed SNP was coded
as an expected allele count. Again, significant associations are defined by a false
discovery rate (FDR) of less than 0.1.
Regulatory profiling of normal ovarian cancer precursor tissues
We performed genome-wide formaldehyde assisted regulatory element (FAIRE) and ChIP
seq with histone 3 lysine 27 acetylation (H3K27ac) and histone 3 lysine 4 monomethylation
(H3K4me) for two normal OSECs, two normal FTSECs and two HGSOC cell lines (UWB1.289
and CAOV3) [Shen et al. in preparation]. These datasets annotate epigenetic signatures
of open chromatin, and collectively indicate transcriptional enhancer regions. We
analysed the FAIRE-seq and ChIP-seq datasets and publically available genomic data
on promoter and UTR domains, intron/exon boundaries, and positions of non-coding RNA
transcripts to identify SNPs from the 100:1 likely causal set that align with biofeatures
that may provide evidence of SNP functionality.
Candidate Gene Analysis Using Genome Wide Profiling of Primary Ovarian Cancers
Data Sets
The Cancer Genome Atlas (TCGA) Project and COSMIC Datasets
TCGA has performed extensive genomic analysis of tumours from a large number of tissue
types including almost 500 high-grade serous ovarian tumours. These data include somatic
mutations, DNA copy number, mRNA and miRNA expression and DNA methylation. COSMIC
is the catalogue of somatic mutations in cancer that collates information on mutations
in tumours from the published literature
59
. They have also identified The Cancer Gene Census, which is a list of genes known
to be involved in cancer. Data are available on a large number of tissue types, including
2,809 epithelial ovarian tumours.
Somatic coding sequence mutations
We analysed all genes for coding somatic sequence mutations generated from either
whole exome or whole genome sequencing. In TCGA, whole exome sequencing data were
available for 316 high-grade serous EOC cases. In addition, we determined whether
mutations had been reported in COSMIC
59
and whether the gene was a known cancer gene in the Sanger Cancer Gene Census.
mRNA expression in tumour and normal tissue
Normalized and gene expression values (Level 3) gene expression profiling data were
obtain from the TCGA data portal for three different platforms (Agilent, Affymetrix
HuEx and Affymetrix U133A). We analysed only the 489 primary serous ovarian tumour
samples included in the final clustering analysis
58
and eight normal fallopian tube samples. The boxplot function in R was used to compare
ovarian tumour samples to the fallopian tube for 91 coding genes with expression data
on any platform within a 1MB region around the most significant SNP at the six loci.
A difference in relative expression between EOC and normal tissue was carried out
using the Wilcoxon rank-sum test.
DNA copy number analysis
Serous EOC samples for 481 tumours with log2 copy number data were analysed using
the cBio portal for analysis of TCGA data
60,61
. For each gene in a region the classes of copy number; homozygous deletion, heterozygous
loss, diploid, gain, and amplification were queried individually using the advanced
onco query language (OQL) option. The frequency of gain and amplification were combined
as “gain”, and homozygous deletion and heterozygous loss were combined as “loss”.
Analysis of copy number vs mRNA expression
Serous EOC samples for 316 complete tumours (those with CNA, mRNA and sequencing data)
were analysed. Graphs were generated using the cBio portal for analysis of TCGA data
and the setting were mRNA expression data Z-score (all genes) with the Z-score threshold
of 2 (default setting) and putative copy number alterations (GISTIC). The Z-score
is the number of standard deviations away from the mean of expression in the reference
population. GISTIC is an algorithm that attempts to identify significantly altered
regions of amplification or deletion across sets of patients.
Luciferase Reporter Assay
The putative causal SNPs at the 1p36 locus lie in the WNT4 promoter and so we tested
their effect on transcription in a luciferase reporter assay (Fig. 2D). Wild-type
and risk haplotype (comprising five correlated variants) sequences corresponding to
the region bound by hg19 co-ordinates chr1:22469416-22470869 were generated by Custom
Gene Synthesis (GenScript Corporation), and then sub-cloned into pGL3-basic (Promega).
Equimolar amounts of luciferase constructs (800 ng) and pRL-TK Renilla (50 ng) were
co-transfected into ~8 × 104 iOSE4
62
normal ovarian cells in triplicate wells of 24 well plates using LipoFectamine 2000
(Life Technologies). Independent transfections were repeated three times. The Dual-Glo
Luciferase Assay kit (Promega) was used to assay luciferase activity 24 hours post
transfection using a BioTek Synergy H4 plate reader. The iOSE-4 cell line (derived
by K. Lawrenson) was maintained under standard conditions and routinely tested for
Mycoplasma and short tandem repeat profiled.
Supplementary Material
1
2
Related collections
Most cited references70
- Record: found
- Abstract: found
- Article: not found
Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal.
J. Gao, B. A. Aksoy, U Dogrusoz … (2015)
- Record: found
- Abstract: found
- Article: not found
The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data.
Ethan Cerami, Jianjiong Gao, Ugur Dogrusoz … (2012)
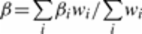
- Record: found
- Abstract: found
- Article: found
METAL: fast and efficient meta-analysis of genomewide association scans
Cristen Willer, Yun Li, Gonçalo R Abecasis (2010)