- Record: found
- Abstract: found
- Article: not found
Cytokine Storm
review-article
Dan L. Longo , M.D.
03 December 2020
Keyword part (code): 2Keyword part (keyword): Hematology/OncologyKeyword part (code): 2_1Keyword part (keyword): Hematology/Oncology General ,
2,
Hematology/Oncology,
Keyword part (code): 2_1Keyword part (keyword): Hematology/Oncology General,
2_1,
Hematology/Oncology General,
Keyword part (code): 10Keyword part (keyword): Emergency MedicineKeyword part (code): 10_6Keyword part (keyword): Shock ,
10,
Emergency Medicine,
Keyword part (code): 10_6Keyword part (keyword): Shock,
10_6,
Shock,
Keyword part (code): 12Keyword part (keyword): Pulmonary/Critical CareKeyword part (code): 12_1Keyword part (keyword): Pulmonary/Critical Care GeneralKeyword part (code): 12_6Keyword part (keyword): Critical Care ,
12,
Pulmonary/Critical Care,
Keyword part (code): 12_1Keyword part (keyword): Pulmonary/Critical Care GeneralKeyword part (code): 12_6Keyword part (keyword): Critical Care ,
12_1,
Pulmonary/Critical Care General,
12_6,
Critical Care,
Keyword part (code): 18Keyword part (keyword): Infectious DiseaseKeyword part (code): 18_1Keyword part (keyword): Infectious Disease General ,
18,
Infectious Disease,
Keyword part (code): 18_1Keyword part (keyword): Infectious Disease General,
18_1,
Infectious Disease General,
Keyword part (code): 19Keyword part (keyword): Allergy/ImmunologyKeyword part (code): 19_1Keyword part (keyword): Allergy/Immunology General ,
19,
Allergy/Immunology,
Keyword part (code): 19_1Keyword part (keyword): Allergy/Immunology General,
19_1,
Allergy/Immunology General
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic has reminded
us of the critical role of an effective host immune response and the devastating effect
of immune dysregulation. This year marks 10 years since the first description of a
cytokine storm that developed after chimeric antigen receptor (CAR) T-cell therapy
1
and 27 years since the term was first used in the literature to describe the engraftment
syndrome of acute graft-versus-host disease after allogeneic hematopoietic stem-cell
transplantation.
2
The term “cytokine release syndrome” was coined to describe a similar syndrome after
infusion of muromonab-CD3 (OKT3).
3
Cytokine storm and cytokine release syndrome are life-threatening systemic inflammatory
syndromes involving elevated levels of circulating cytokines and immune-cell hyperactivation
that can be triggered by various therapies, pathogens, cancers, autoimmune conditions,
and monogenic disorders.
From a historical perspective, cytokine storm was previously referred to as an influenza-like
syndrome that occurred after systemic infections such as sepsis and after immunotherapies
such as Coley’s toxins.
4
Yersinia pestis infection (i.e., the plague) has led to major pandemics (e.g., the
Black Death) and triggers alveolar macrophages to produce excessive amounts of cytokines,
resulting in cytokine storm.
5
An exaggerated immune response was suspected to contribute to the lethality of the
1918–1919 influenza pandemic. In fact, a reconstructed H1N1 virus isolated from the
1918 pandemic, as compared with common reference strains of the virus that causes
influenza A, triggered marked pulmonary inflammation in mice.
6
Recognition that the immune response to the pathogen, but not the pathogen itself,
can contribute to multiorgan dysfunction and that similar cytokine storm syndromes
could occur with no obvious infection led to the investigation of immunomodulators
and cytokine-directed therapies. One of the earliest targeted therapies for abrogation
of a cytokine storm was the anti–interleukin-6 receptor monoclonal antibody tocilizumab,
which was developed for the treatment of idiopathic multicentric Castleman’s disease
in the 1990s. A host of other disorders have been described as causes of cytokine
storm and targeted with immune-directed therapies, such as sepsis, primary and secondary
hemophagocytic lymphohistiocytosis (HLH), autoinflammatory disorders, and coronavirus
disease 2019 (Covid-19).
No single definition of cytokine storm or the cytokine release syndrome is widely
accepted, and there is disagreement about how these disorders differ from an appropriate
inflammatory response. The National Cancer Institute’s definition, based on the Common
Terminology Criteria for Adverse Events (CTCAE), is too broad, since the criteria
for an inflammatory syndrome can also apply to other physiological states, and the
definition of the American Society for Transplantation and Cellular Therapy is based
on criteria that focus too specifically on iatrogenic causes of cytokine storm alone.
7
Although cytokine storm is easy to identify in disorders with elevated cytokine levels
in the absence of pathogens, the line between a normal and a dysregulated response
to a severe infection is blurry, especially considering that certain cytokines may
be both helpful in controlling an infection and harmful to the host. The interdependence
of these inflammatory mediators further complicates the distinction between a normal
and a dysregulated response.
It is important for the clinician to recognize cytokine storm because it has prognostic
and therapeutic implications. In this review, we propose a unifying definition of
cytokine storm; discuss the pathophysiological features, clinical presentation, and
management of the syndrome; and provide an overview of iatrogenic, pathogen-induced,
neoplasia-induced, and monogenic causes. Our goal is to provide physicians with a
conceptual framework, a unifying definition, and essential staging, assessment, and
therapeutic tools to manage cytokine storm.
Clinical Features and Laboratory Abnormalities
Cytokine storm is an umbrella term encompassing several disorders of immune dysregulation
characterized by constitutional symptoms, systemic inflammation, and multiorgan dysfunction
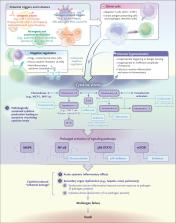
that can lead to multiorgan failure if inadequately treated (Figure 1). The onset
and duration of cytokine storm vary, depending on the cause and treatments administered.
7
Although the initial drivers may differ, late-stage clinical manifestations of cytokine
storm converge and often overlap. Nearly all patients with cytokine storm are febrile,
and the fever may be high grade in severe cases.
8
In addition, patients may have fatigue, anorexia, headache, rash, diarrhea, arthralgia,
myalgia, and neuropsychiatric findings. These symptoms may be due directly to cytokine-induced
tissue damage or acute-phase physiological changes or may result from immune-cell–mediated
responses. Cases can progress rapidly to disseminated intravascular coagulation with
either vascular occlusion or catastrophic hemorrhages, dyspnea, hypoxemia, hypotension,
hemostatic imbalance, vasodilatory shock, and death. Many patients have respiratory
symptoms, including cough and tachypnea, that can progress to acute respiratory distress
syndrome (ARDS), with hypoxemia that may require mechanical ventilation. The combination
of hyperinflammation, coagulopathy, and low platelet counts places patients with cytokine
storm at high risk for spontaneous hemorrhage.
In severe cases of cytokine storm, renal failure, acute liver injury or cholestasis,
and a stress-related or takotsubo-like cardiomyopathy can also develop.
9
The combination of renal dysfunction, endothelial-cell death, and acute-phase hypoalbuminemia
can lead to capillary leak syndrome and anasarca — changes that are similar to those
observed in patients with cancer who are treated with high-dose interleukin-2.
10
Neurologic toxicity associated with T-cell immunotherapy is referred to as immune
effector cell–associated neurotoxicity syndrome or cytokine release syndrome–associated
encephalopathy.
7
The neurologic toxic effects are often delayed, developing several days after the
onset of the cytokine storm.
The laboratory findings in cytokine storm are variable and influenced by the underlying
cause. Nonspecific markers of inflammation such as C-reactive protein (CRP) are universally
elevated and correlate with severity.
11
Many patients have hypertriglyceridemia and various blood-count abnormalities, such
as leukocytosis, leukopenia, anemia, thrombocytopenia, and elevated ferritin and d-dimer
levels. Changes in circulating cell counts are most likely due to a complex interplay
among cytokine-induced changes in production and mobilization of cells from the bone
marrow, immune-mediated destruction, and chemokine-induced migration. Prominent elevations
in serum inflammatory cytokine levels, such as interferon-γ (or CXCL9 and CXCL10,
chemokines induced by interferon-γ), interleukin-6, interleukin-10, and soluble interleukin-2
receptor alpha, a marker of T-cell activation, are usually present. Highly elevated
serum interleukin-6 levels are found in CAR T-cell therapy–induced cytokine storm
and several other cytokine storm disorders.
8
The approach to evaluating a patient with cytokine storm should accomplish the following
three main goals: identifying the underlying disorder (and ruling out disorders that
may mimic cytokine storm), establishing severity, and determining the clinical trajectory.
A complete workup for infection, as well as laboratory assessment of kidney and liver
function, should be performed in all suspected cases of cytokine storm. Measurements
of inflammatory acute-phase biomarkers, such as CRP and ferritin, and blood counts
should be obtained, since they correlate with disease activity. Arterial blood-gas
measurement should be performed if the respiratory evaluation warrants it. Cytokine
profiles may be helpful in determining the trend from baseline values, although these
findings are typically not available soon enough to include as part of the immediate
workup or to guide treatment decisions.
Establishing the disorder underlying the cytokine storm can be challenging. Cytokine
storm is not a diagnosis of exclusion, and it can encompass many disorders. For example,
patients may have both sepsis and cytokine storm. However, it is important to distinguish
between cytokine storm due to an iatrogenic cause such as CAR T-cell therapy and cytokine
storm due to systemic infection, since immunosuppressive treatments could be detrimental
if used in patients with septicemia. Unfortunately, it is difficult to distinguish
cytokine storm due to sepsis from cytokine storm due to CAR T-cell therapy on the
basis of clinical features alone. Levels of serum cytokines — most prominently, interferon-γ
— are often more elevated in patients with cytokine storm due to CAR T-cell therapy
than in patients with sepsis-induced cytokine storm, who often have higher levels
of circulating interleukin-1β, procalcitonin, and markers of endothelial damage.
12
Thus, combinations of assays to rule out infection and measure serum cytokines can
help to identify the cause of the cytokine storm. However, CAR T-cell therapy and
other noninfectious causes can also occur with infections, and infections can develop
during the course of therapy, so continued monitoring for infections is warranted.
Disorders that should be ruled out in considering cytokine storm include anaphylaxis
and physiological responses to microbial infections.
The grading systems used to predict and assess the severity of cytokine storm differ
according to the cause. Serum biomarkers, including glycoprotein 130 (gp130), interferon-γ,
and interleukin-1–receptor antagonist (IL1RA), can be used to predict the severity
of cytokine storm induced by CAR T-cell therapy,
13
with a separate grading scale used to assess the current severity.
7
HScore and MS score are used for classifying HLH-associated cytokine storm, and HLH-2004
guides treatment. For the grading of cytokine storm due to other causes, the immune
systems disorders section of CTCAE is used (https://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_5x7.pdf).
Pathophysiological Features of Cytokine Storm
Inflammation involves a set of biologic mechanisms that evolved in multicellular organisms
to contain invasive pathogens and resolve injuries by activating innate and adaptive
immune responses. The immune system is expected to recognize foreign invaders, respond
proportionally to the pathogen burden, and then return to homeostasis. This response
requires a balance between sufficient cytokine production to eliminate the pathogen
and avoidance of a hyperinflammatory response in which an overabundance of cytokines
causes clinically significant collateral damage. Cytokines play a key role in coordinating
antimicrobial effector cells and providing regulatory signals that direct, amplify,
and resolve the immune response. Cytokines have short half-lives, which normally prevents
them from having effects outside lymphoid tissue and sites of inflammation. Although
typically considered to be pathologic, sustained production of cytokines that leads
to elevated circulating levels may be necessary to appropriately control some disseminated
infections. At increased levels, cytokines can have systemic effects and cause collateral
damage to vital organ systems.
Immune hyperactivation in cytokine storm can occur as a result of inappropriate triggering
or danger sensing, with a response initiated in the absence of a pathogen (e.g., in
genetic disorders involving inappropriate inflammasome activation or idiopathic multicentric
Castleman’s disease); an inappropriate or ineffective amplitude of response, involving
excessive effector immune-cell activation (e.g., in cytokine storm due to CAR T-cell
therapy), an overwhelming pathogen burden (e.g., in sepsis), or uncontrolled infections
and prolonged immune activation (e.g., in HLH associated with Epstein–Barr virus [EBV]);
or failure to resolve the immune response and return to homeostasis (e.g., in primary
HLH) (Figure 2). In each of these states, there is a failure of negative feedback
mechanisms that are meant to prevent hyperinflammation and the overproduction of inflammatory
cytokines and soluble mediators. The excessive cytokine production leads to hyperinflammation
and multiorgan failure. Regulatory cell types, decoy receptors for proinflammatory
cytokines such as IL1RA, and antiinflammatory cytokines such as interleukin-10 are
important for antagonizing inflammatory-cell populations and preventing immune hyperactivity.
Given the lack of a unifying definition for cytokine storm
14
and disagreement about the distinction between cytokine storm and a physiologic inflammatory
response, we propose the following three criteria for identifying cytokine storm:
elevated circulating cytokine levels, acute systemic inflammatory symptoms, and either
secondary organ dysfunction (often renal, hepatic, or pulmonary) due to inflammation
beyond that which could be attributed to a normal response to a pathogen (if a pathogen
is present), or any cytokine-driven organ dysfunction (if no pathogen is present).
Improvement in outcomes with cytokine neutralization or antiinflammatory agents further
supports the pathologic role of excessive cytokines and the classification of a condition
as a cytokine storm. However, lack of a treatment response does not necessarily rule
out cytokine storm, because underlying conditions are likely to play a part, a different
cytokine may be the disease driver, or the timing of treatment may have been poor.
In short, cytokine storm involves an immune response that causes collateral damage,
which may be greater than the immediate benefit of the immune response. Thus, an exuberant
inflammatory response to a large pathogen burden may be appropriate for controlling
the infection if excessive secondary organ dysfunction does not occur, whereas similarly
high levels of cytokines in cancer-associated HLH or idiopathic multicentric Castleman’s
disease would be considered a pathologic state of cytokine storm because no pathogen
requiring an immune response is involved and patients benefit from treatment with
cytokine neutralization and other antiinflammatory agents. Circulating cytokine levels
can be difficult to measure because cytokines have short half-lives, circulating levels
may not accurately reflect local tissue levels, and measurements may not be easily
obtained worldwide. We do not propose a specific threshold for elevations in cytokine
levels above the normal range, and we do not recommend specific cytokine panels or
list particular cytokines whose levels must be elevated, given the lack of available
evidence. However, we believe that this is an important area for future research and
could benefit from systematic assessment by a multidisciplinary consortium.
Cell Types Involved in Cytokine Storm
The cells of the innate immune system are the first line of defense against pathogens.
Neutrophils, monocytes, and macrophages recognize pathogens, produce cytokines, and
engulf pathogens and cells by phagocytosis. There are many other innate immune cells,
such as dendritic cells, gamma–delta T cells, and natural killer (NK) cells.
15
Innate immune cells use pattern-recognition receptors, which are not specific for
any particular antigen, to recognize and respond to a wide variety of microbial invaders
by producing cytokines that activate cells of the adaptive immune system.
Innate cells that are most often implicated in the pathogenesis of cytokine storm
include neutrophils, macrophages, and NK cells. Neutrophils can produce neutrophil
extracellular traps, a network of fibers that contribute to thrombi formation and
amplify cytokine production during cytokine storm. Macrophages, which are tissue-resident
cells that are often derived from circulating monocytes, do not divide; they have
diverse functions, from the removal of senescent cells by engulfment, to tissue repair
and immunoregulation, to antigen presentation. In many forms of cytokine storm, macrophages
become activated and secrete excessive amounts of cytokines, ultimately causing severe
tissue damage that can lead to organ failure. Hemophagocytic macrophages are often
observed in bone marrow biopsy specimens from patients with cytokine storm. Interferon-γ
can induce hemophagocytosis by macrophages, and this may contribute to the cytopenias
commonly observed in patients with cytokine storm.
16
The cytolytic function of NK cells is diminished in some forms of cytokine storm,
which can lead to prolonged antigenic stimulation and difficulty resolving inflammation.
17
Excess interleukin-6 may mediate the impairment in NK-cell function by lowering perforin
and granzyme production.
The adaptive immune system is composed of B cells and T cells. T cells differentiate
into a number of subsets with distinct effector-cell functions potentially involved
in cytokine storm (Figure 3). Type 1 helper T (Th1) cells and cytotoxic T lymphocytes
(CTLs) are primarily responsible for the host defense against viral infections. Th1
cells regulate the recruitment of macrophages, whereas type 2 helper T (Th2) cells
recruit eosinophils and basophils, type 9 helper T (Th9) cells recruit mast cells,
and type 17 helper T (Th17) cells recruit neutrophils.
18
An exaggerated Th1-type inflammatory response often occurs during cytokine storm.
Th1 cells produce large quantities of interferon-γ, induce delayed hypersensitivity
reactions, activate macrophages, and are essential for defense against intracellular
pathogens.
19
Iatrogenic causes of cytokine storm involving excessive T-cell activation, such as
CAR T-cell and anti-CD28 antibody therapy, point to the ability of activated T cells
to initiate cytokine storm. Impaired granule-mediated killing of infected cells or
tumor cells by CTLs is a key aspect of some forms of cytokine storm.
20
Data from mouse models of HLH and patients with cytokine storm indicate that the inability
of CTLs to kill efficiently leads to prolonged activation of T cells, triggering a
cascade of inflammatory tissue damage.
21-23
Th17 cells have a major role in host defense, particularly antifungal protection,
and abnormal Th17-cell function can lead to autoimmunity.
24
An experimental model of macrophage activation syndrome (a form of secondary HLH)
provides evidence that Th17 cells can be drivers of a cytokine storm that is independent
of interferon-γ.
25
B cells are not often associated with the pathogenesis of cytokine storm. However,
the effectiveness of B-cell depletion in treating some cytokine storm disorders, such
as human herpesvirus 8 (HHV-8)–associated multicentric Castleman’s disease, suggests
that these cells are capable of initiating or propagating cytokine storm, particularly
when virally infected.
Cytokines
As noted above, the recognition of cytokine storm as an entity is relatively recent.
The advent of molecular cloning technologies led to the discovery of the panoply of
cytokines and chemokines involved in cytokine storm (Table 1); the realization that
diverse entities can cause cytokine storm (Table 2) also contributed to its recognition.
The administration of recombinant cytokines (e.g., interleukin-1, interleukin-6, interleukin-12,
interleukin-18, tumor necrosis factor [TNF], and interferon-γ) in animal models and
for cancer treatment in humans induces severe toxic effects or lethality consistent
with the central role of cytokines as mediators of hyperinflammation in cytokine storm.
27-29
Conversely, reduction in symptoms and improvement in organ function with neutralization
of specific cytokines with monoclonal antibodies also reveal that excessive levels
of certain cytokines play a critical role in a number of cytokine storm disorders.
A complex, interconnected network of cell types, signaling pathways, and cytokines
is involved in cytokine storm disorders. Interferon-γ, interleukin-1, interleukin-6,
TNF, and interleukin-18 are key cytokines that often have elevated levels in cytokine
storm and are thought to have central immunopathologic roles. The pattern of cytokine
elevations varies on the basis of such factors as the microbiome, genetic features,
and underlying disorders.
30
The specific immune cells that secrete the various cytokines are not fully understood
and most likely vary among cytokine storm disorders. Interferon-γ is primarily secreted
by activated T cells and NK cells and is a potent activator of macrophages. Clinically,
interferon-γ causes fever, chills, headache, dizziness, and fatigue.
31
Emapalumab, a monoclonal antibody that binds interferon-γ, was recently approved for
the treatment of cytokine storm in patients with primary HLH.
32
This agent may also be useful in other cytokine storm disorders, such as macrophage
activation syndrome or CAR T-cell–associated cytokine storm, although in the latter
case, it may diminish antitumor effects.
Fever, a clinical hallmark of cytokine storm, can be elicited by interleukin-1, interleukin-6,
or TNF through distinct mechanisms. Interleukin-1 is encoded by two genes (IL1A and
IL1B), both of which bind to the same interleukin-1 receptor, activating a cascade
of intracellular signaling pathways, including nuclear factor κB (NF-κB). The interleukin-1–receptor
antagonist anakinra is effective as a single agent and in combination with other agents
for the treatment of some forms of cytokine storm.
33,34
Levels of interleukin-6, an important mediator of the acute inflammatory response
and pathophysiological features of cytokine storm, are highly elevated across various
underlying immunopathologic disorders
35,36
and in mouse models of cytokine storm.
37
Both tocilizumab, a monoclonal antibody directed at the interleukin-6 receptor (interleukin-6R),
and siltuximab, which neutralizes interleukin-6 directly, have been shown to be effective
in a number of cytokine storm disorders, including HLH, idiopathic multicentric Castleman’s
disease, and CAR T-cell–induced cytokine storm.
38
Interleukin-6 is one of the more complex cytokines, since it is produced by and acts
on immune and nonimmune cells across multiple organ systems. It can signal through
two main pathways, referred to as classic cis signaling and trans signaling.
38
The membrane-bound interleukin-6R does not possess intracellular signaling domains
but signals instead through interaction with membrane-bound gp130. In cis signaling,
soluble interleukin-6 binds to membrane-bound interleukin-6R, forming an interleukin-6–interleukin-6R
complex that binds to gp130, which then initiates signaling through its intracellular
domain.
Downstream signal transduction is mediated by JAKs (Janus kinases) and STAT3 (signal
transducer and activator of transcription 3), as well as by Akt–mTOR (mammalian target
of rapamycin) and MAPK–ERK (mitogen-activated protein kinase–extracellular signal-regulated
kinase) pathways. Membrane-bound gp130 is ubiquitously expressed, whereas expression
of membrane-bound interleukin-6R is restricted largely to immune cells. Activation
of cis signaling results in pleiotropic effects on the immune system, which can contribute
to cytokine storm.
38
In the presence of high circulating levels of interleukin-6, which can be present
in cytokine storm, trans signaling occurs through the binding of interleukin-6 to
the soluble form of interleukin-6R, forming a complex with a gp130 dimer on potentially
all cell surfaces. The resultant interleukin-6–soluble interleukin-6R–gp130–JAK-STAT3
signaling is then activated in cells that do not express the membrane-bound interleukin-6R,
such as endothelial cells. This results in systemic hyperinflammation involving secretion
of monocyte chemoattractant protein 1 (MCP-1), interleukin-8, and additional interleukin-6,
as well as increased vascular endothelial growth factor (VEGF) and reduced E-cadherin
expression on endothelial cells, which contribute to vascular hyperpermeability, leakiness,
hypotension, and pulmonary dysfunction.
38
TNF is a potent, multifunctional, proinflammatory cytokine that belongs to the TNF–TNF
receptor superfamily. In addition to inducing fever, augmenting systemic inflammation,
and activating antimicrobial responses such as interleukin-6, TNF can induce cellular
apoptosis and regulate immunity. TNF and other cytokines in the TNF–TNF receptor superfamily
are potent inducers of NF-κB, leading to the expression of multiple proinflammatory
genes. In mouse models of toxic shock, TNF is the cytokine driver of superantigen-driven
cytokine storm.
39
The effectiveness of anti-TNF therapies in certain autoinflammatory-driven cytokine
storm conditions points to their potential role in the treatment of cytokine storm,
but the limitations and dangers of anti-TNF therapies in patients with sepsis indicate
that more work is needed.
Interleukin-18 is a member of the large interleukin-1 family
40
that has recently been associated with cytokine storm disorders. Interleukin-18 and
interleukin-1β are activated from precursors by inflammasomes. The inflammasome is
a multimolecular cytosolic sensor that detects pathogenic microorganisms and sterile
stressors and activates caspase-1 during the process of pyroptosis, which, in turn,
causes the inactive precursor forms of interleukin-1β and interleukin-18 to become
the active forms.
41,42
Macrophages and dendritic cells are the primary sources of bioactive interleukin-18,
which has many proinflammatory effects. Most important, it synergizes with interleukin-12
or interleukin-15 to stimulate secretion of interferon-γ from T cells and NK cells,
and thus promotes Th1-type inflammatory responses. The interleukin-18 receptor is
constitutively expressed on NK cells and induced on activation in most T cells. Interleukin-1β
and interleukin-18 are also potent inducers of interleukin-6 secretion from macrophages.
43
Patients with cytokine storm due to macrophage activation syndrome have high levels
of interleukin-18 in serum,
44
and interleukin-18 is a biomarker of severity that correlates with hyperferritinemia,
elevated aminotransferase levels, and disease flare.
45
The proinflammatory effects of interleukin-18 are normally kept in check by the interleukin-18–binding
protein (IL18BP), which prevents the binding of interleukin-18 to its receptor.
46
The ratio of free interleukin-18 to bound interleukin-18–IL18BP complexes in serum
is an important indicator of the severity of the macrophage activation syndrome.
44,47
Tadekinig alfa is a recombinant IL18BP currently under investigation as a treatment
for hyperinflammation.
Chemokines are a class of cytokines that contribute to a variety of immune-cell functions,
including leukocyte recruitment and trafficking. Dysregulated trafficking during inflammation
may have a role in hyperinflammation. Numerous regulatory cytokines such as interleukin-10
and natural cytokine antagonists such as IL1RA serve as buffers to limit systemic
off-target effects. Interleukin-10 inhibits the production of TNF, interleukin-1,
interleukin-6, and interleukin-12 and down-regulates antigen presentation. Furthermore,
in mice lacking interleukin-10, infection leads to cytokine storm.
48
Though interleukin-10 and IL1RA are often elevated in cytokine storm, this finding
most likely reflects a secondary, albeit insufficient, counterregulatory response
to the proinflammatory cytokines. Anakinra is a therapeutic agent that mimics the
endogenous immunoregulatory effects of IL1RA.
Plasma proteins such as complement proteins and other inflammatory mediators can contribute
to the pathogenesis of cytokine storm. These soluble proteins recognize pathogens,
amplify cellular responses, and provide feedback on cytokine signaling. In fact, cytokines
can enhance the production of complement proteins, which in turn can enhance or inhibit
cytokine production. Thus, complement can be highly effective in eliminating microbes
but can also cause collateral damage if excessive. Hypocomplementemia, resulting from
increased consumption by immune complexes, can be observed in cytokine storm.
49
Complement inhibitors are under evaluation for the treatment of cytokine storm disorders.
Iatrogenic Cytokine Storm
Infusion of CAR T cells engineered to recognize and eliminate CD19+ lymphoma cells
can induce cytokine storm, with supraphysiologic levels of interferon-γ and interleukin-6.
50
The highly activated CAR T cells are clearly the initiators of the cytokine storm.
Although some studies suggest that the driver cytokines are released by CAR T cells,
resulting in a positive feedback loop of T-cell activation and inflammatory cytokine
release,
51
recent studies in mice suggest that the cytokines and factors mediating the severity
of cytokine storm are produced not by the CAR T cells but by macrophages and can be
reversed by interleukin-6 and interleukin-1 blockade.
52-54
Tumor lysis most likely also contributes to the cytokine storm through the induction
of pyroptosis in target cells.
55
Since interleukin-6 blockade is highly effective at reversing symptoms and organ dysfunction
in most patients, it is the likely cytokine driver of cytokine storm induced by CAR
T-cell therapy. Glucocorticoids and interleukin-1 inhibition can also be effective
in the treatment of this type of cytokine storm.
Cytokine storm can be observed with other T-cell–engaging immunotherapies as well,
such as blinatumomab, a bispecific antibody that binds to CD19+ and CD3+ T cells.
56
Like CAR T cells, activated T cells initiate the cytokine storm, and macrophage activation
propagates blinatumomab-induced cytokine storm, which also responds to anti–interleukin-6
antibody therapy.
36
The unfortunate consequences of another T-cell–activating treatment with the anti-CD28
superagonist TGN1412 show that rapid activation of large numbers of T cells can result
in severe cytokine storm within minutes after infusion.
57
However, cytokine storm does not develop in all patients treated with CAR T cells
or blinatumomab, so additional factors, such as CAR structure and design,
51
disease burden,
58
and host genomic background,
59
are likely to play a part. In a recent study of NK-cell CAR therapy, there were no
reported cases of cytokine storm or even elevated interleukin-6 levels,
60
possibly because of lower interleukin-6 production by NK cells than by T cells and
different cross-talk with myeloid cells. Additional iatrogenic causes of cytokine
storm include rituximab,
35
gene therapies, immune checkpoint inhibitors, cardiac-bypass surgery,
61
and allogeneic stem-cell transplantation, as well as bioterrorism agents such as staphylococcal
enterotoxin B and Francisella tularensis.
Pathogen-Induced Cytokine Storm
Cytokine storm can also result from naturally occurring microbial infections. Though
data on relative frequencies are limited, infections are most likely the most common
trigger of cytokine storm. Distinguishing between appropriate cytokine production
for controlling a widespread infection and excessive cytokine production is challenging.
Disseminated bacterial infections causing sepsis induce the production of many cytokines
that can lead to fever, cell death, coagulopathies, and multiorgan dysfunction. The
collateral damage caused by the immune response as it attempts to clear the pathogen
can be more deadly than the pathogen itself. Certain bacteria, including streptococcus
species and Staphylococcus aureus, can produce superantigens that cross-link the major
histocompatibility complex and T-cell receptors, leading to polyclonal activation
of T cells, cytokine production, and toxic shock syndrome. Superantigens are the most
powerful T-cell mitogens, and bacterial superantigen concentrations of less than 0.1
pg per milliliter are sufficient to stimulate T cells in an uncontrolled manner, resulting
in fever, shock, and death.
In sepsis-associated cytokine storm, it is unclear which immune cell types and cytokines
may be responsible for propagating the pathologic hyperinflammation. Antibiotics are
the mainstay of treatment. The administration of monoclonal antibodies directed at
specific cytokines and the use of apheresis or medical devices to remove cytokines
from circulation have had generally disappointing results in clinical trials.
62
Although the timing of treatment in these studies may have contributed to the lack
of benefit, additional host or pathogen factors may be important, beyond the specifically
elevated cytokine levels. For example, reanalysis of a negative trial of interleukin-1β
blockade in patients with sepsis identified a subgroup of patients with elevated ferritin
levels who seemed to benefit from the treatment.
63
Disseminated viral infections can also induce profound cytokine storm. Patients with
hyperinflammatory responses to microbes often have defects in pathogen detection,
effector and regulatory mechanisms, or resolution of inflammation. For example, patients
lacking functional perforin, which is critical for resolving infections and inflammation,
have prolonged CD8+ T-cell production of interferon-γ and TNF, and HLH-associated
cytokine storm develops in such patients when they are infected with EBV or cytomegalovirus.
64
Experimental models suggest that cytokine storm occurs in these patients from defective
perforin-mediated cytolysis that leads to prolonged engagement between lymphocytes
and antigen-presenting cells and defective clearance of antigen-bearing dendritic
cells, resulting in continuous activation and proliferation of T cells and macrophages,
hemophagocytosis, and an autocrine loop of proinflammatory cytokines.
21,65-67
Furthermore, retrospective analyses of data from persons who died from coagulopathies
and hemophagocytosis during the H1N1 influenza pandemic of 2009 revealed germline
mutations previously associated with HLH-associated cytokine storm.
30
Thus, the pathogen initiates and T-cell activation propagates cytokine storm in patients
with a genetic susceptibility. Cyclosporine and anti–interleukin-6 receptor monoclonal
antibody therapy can be effective in some virus-driven forms of HLH-associated cytokine
storm, indicating the critical role of T-cell activation and interleukin-6.
Another pathogen-induced form of cytokine storm is HHV-8–associated multicentric Castleman’s
disease. In this disorder, uncontrolled infection with HHV-8 (also known as Kaposi’s
sarcoma herpesvirus) leads to a cytokine storm driven primarily by excessive production
of human interleukin-6 and viral interleukin-6 by HHV-8–infected plasmablasts.
68
Patients with HHV-8–associated multicentric Castleman’s disease are immunocompromised
as a result of human immunodeficiency virus infection or a genetic susceptibility,
making it difficult to control the HHV-8 infection, which is a common, typically asymptomatic
infection in the general population.
69
A recent study showed that the effect of tocilizumab in patients with HHV-8–associated
multicentric Castleman’s disease was minimal and short-lived, most likely because
of viral interleukin-6 signaling that was independent of the neutralized interleukin-6
receptor.
70
As with EBV-associated HLH,
71
rituximab is highly effective in patients with HHV-8–associated multicentric Castleman’s
disease, since B-cell depletion removes the primary reservoir for HHV-8.
72
Many additional microbes can trigger cytokine storm, including other herpesviruses,
such as herpes simplex virus, and other influenza viruses, such as H5N1.
Targeted treatment is more challenging in patients with viral infections than in patients
with bacterial infections, since fewer antiviral agents are available. Intravenous
immune globulin and convalescent plasma are sometimes used to help control the pathogen
and provide beneficial immunomodulation. For some viral infections, treating patients
with proinflammatory cytokines in the early stages of infection can help to control
the virus before detrimental effects of the immune response occur.
73
Monogenic or Autoimmune Cytokine Storm
In rare cases, a pathogen triggers cytokine storm in patients with monogenic disorders,
and in other cases, cytokine storm has autoimmune, neoplastic, or idiopathic causes.
In patients with primary HLH, various autosomal recessive monogenic abnormalities
in granule-mediated cytotoxicity lead to cytokine storm. Common pathologic mutations
include those occurring in PRF1, UNC13D, STXBP1, RAB27A, STX11, SH2D1A, XIAP, and
NLRC4.
23
In patients with secondary HLH, viral, autoimmune, or neoplastic disorders trigger
cytokine storm, and such patients often have heterozygous polymorphisms in the same
genes that are altered in primary HLH.
65,74
Elevated levels of interferon-γ, TNF, interleukin-1, interleukin-4, interleukin-6,
interleukin-8, interleukin-10, CXCL9, CXCL10, and interleukin-18 are frequently associated
with HLH. Anti–interferon-γ antibody therapy with emapalumab has recently been approved
for the treatment of primary HLH, as a bridge to allogeneic stem-cell transplantation,
which is typically curative.
The beneficial effects of glucocorticoids, cyclosporine, anti–interleukin-1 antibody,
JAK1 and JAK2 inhibitors, anti–interleukin-6 antibody, and cytotoxic chemotherapies
in some patients with primary or secondary HLH suggest that pathways targeted by these
agents are key to pathogenesis. Cyclophosphamide and etoposide, which are broadly
cytotoxic but particularly effective at eliminating activated CD8+ T cells, are often
effective in patients with primary HLH, secondary HLH (including macrophage activation
syndrome), and corresponding models.
75
Etoposide also targets macrophages, including those involved in regulating inflammation,
which could be harmful. Generalized T-cell and B-cell ablation with alemtuzumab and
T-cell ablation with antithymocyte globulin have been reported; ablation most likely
works by depleting the pathogenic CD8+ T cells, among other cell types.
76
Nonablative inhibition of T cells with cyclosporine can also be helpful.
77
Autoinflammatory diseases are characterized by seemingly unprovoked inflammation and
cytokine storm without signs of infection or autoimmunity. Affected patients have
germline mutations in genes regulating the innate immune system and activation of
the inflammasome. Several genetic disorders are associated with altered regulation
of the innate immune system, including familial Mediterranean fever (MEFV), TNF receptor–associated
periodic syndrome (TNFRSF1A), hyperimmunoglobulinemia D with periodic fever syndrome
(MVK), familial cold autoinflammatory syndrome (NLRP3), the Muckle–Wells syndrome
(NLRP3), neonatal-onset multisystem inflammatory disease (NLRP3), deficiency of ADA2
(CECR1), NLRC4 inflammasomopathies, X-linked lymphoproliferative type 2 disorder (XIAP),
the Takenouchi–Kosaki syndrome (CDC42), and the Wiskott–Aldrich syndrome (CDC42).
Although all patients with these disorders have periodic fevers, only a portion have
cytokine storm. Given the primary genetic defects and the effective treatments that
are available, innate cells are most likely the primary cell drivers involved, and
TNF, interleukin-1, interleukin-18, or a combination of these cytokines probably drives
pathogenesis. Patients with genetic immunodeficiency syndromes such as chronic granulomatous
disease and STAT1 gain-of-function disease can, paradoxically, present with cytokine
storm from overwhelming infections.
78
Idiopathic multicentric Castleman’s disease is another cytokine storm disorder that
is similar to HHV-8–associated multicentric Castleman’s disease, but the cause is
unknown. Patients with the thrombocytopenia, anasarca, fever, reticulin fibrosis,
and organomegaly (TAFRO) subtype tend to have the most severe cytokine storm.
79
Although the cause is unknown, interleukin-6 is the driver of pathogenesis in a large
portion of patients. As a result, tocilizumab, which targets the interleukin-6 receptor,
and siltuximab, which targets interleukin-6 directly, were developed and approved
by regulatory agencies in Japan (tocilizumab) and in the United States and dozens
of other countries (siltuximab) for the treatment of idiopathic multicentric Castleman’s
disease. Both siltuximab and tocilizumab have been shown to resolve disease flares
and sustain remission in approximately one third to one half of patients.
80
However, some patients with low circulating interleukin-6 levels have a response to
interleukin-6 blockade, and some patients with high systemic interleukin-6 levels
do not have a response. A seven-protein panel that can predict which patients with
idiopathic multicentric Castleman’s disease are most likely to benefit from siltuximab
was recently identified and validated (https://ashpublications.org/blood/article/132/Supplement%201/3716/265269/Serum-Proteomics-Reveals-Distinct-Subtypes?searchresult=1).
Patients with idiopathic multicentric Castleman’s disease who have progressive organ
dysfunction and who do not have a response to anti–interleukin-6 therapy are often
treated with combination cytotoxic chemotherapy to nonspecifically eliminate hyperinflammatory
cells.
81
Other elevated serum cytokines and cellular signaling pathways that could be considered
for therapeutic targeting include CXCL13, CXCL10 (interferon-inducible protein 10
[IP-10]), VEGF-A,
82
type I interferon,
83
mTOR complex 1 (mTORC1),
84
and JAK-STAT3. These findings have led to treatment with the mTORC1 inhibitor sirolimus
in patients with idiopathic multicentric Castleman’s disease who do not have a response
to anti–interleukin-6 therapy.
85
Sirolimus therapy is being evaluated in an ongoing clinical trial involving patients
with active disease who do not yet have fulminant cytokine storm (ClinicalTrials.gov
number, NCT03933904).
Covid-19–Associated Cytokine Storm
Covid-19, which is caused by SARS-CoV-2, is characterized by heterogeneous symptoms
ranging from mild fatigue to life-threatening pneumonia, cytokine storm, and multiorgan
failure. Cytokine storm was also reported in patients with SARS and was associated
with poor outcomes.
86
Although the mechanisms of lung injury and multiorgan failure in Covid-19 are still
under investigation,
14
reports of hemophagocytosis and elevated cytokine levels — as well as beneficial effects
of immunosuppressant agents — in affected patients, particularly those who are the
most severely ill, suggest that cytokine storm may contribute to the pathogenesis
of Covid-19.
87,88
Serum cytokine levels that are elevated in patients with Covid-19–associated cytokine
storm include interleukin-1β, interleukin-6, IP-10, TNF, interferon-γ, macrophage
inflammatory protein (MIP) 1α and 1β, and VEGF.
89,90
Higher interleukin-6 levels are strongly associated with shorter survival.
91
The relative frequencies of circulating activated CD4+ and CD8+ T cells and plasmablasts
are increased in Covid-19.
92
In addition to the elevated systemic cytokine levels and activated immune cells, several
clinical and laboratory abnormalities, such as elevated CRP and d-dimer levels, hypoalbuminemia,
renal dysfunction, and effusions, are also observed in Covid-19, as they are in cytokine
storm disorders. Laboratory test results reflecting hyperinflammation and tissue damage
were found to predict worsening outcomes in Covid-19.
93
Although immunologic dysregulation has been observed in severe cases of Covid-19,
26
it is not known whether immune hyperactivity or a failure to resolve the inflammatory
response because of ongoing viral replication or immune dysregulation underlies severe
cases. The correlation between the nasopharyngeal viral load and cytokine levels (e.g.,
interferon-α, interferon-γ, and TNF), as well as a declining viral load in moderate
but not severe cases, suggests that the immune response is positively associated with
the viral burden.
26
Alternatively, the discoveries of inborn errors of type I interferon immunity and
autoantibodies against type I interferons in the most severe cases of Covid-19 suggest
that an inadequate antiviral response may be contributory in some patients with Covid-19.
94,95
Host immune responses and immune-related symptoms are extremely variable between asymptomatic
patients (who have effective control of SARS-CoV-2) and patients with severe Covid-19
(who are unable to control the virus), which suggests that host immune dysregulation
contributes to pathogenesis in some cases. Another hypothesized mechanism involves
autoimmunity due to molecular mimicry between SARS-CoV-2 and a self-antigen. These
mechanisms may be involved in subgroups of patients, such as children with postinfection
multisystem inflammatory syndrome, a condition that seems to be ameliorated by immunomodulatory
therapies such as intravenous immune globulin, glucocorticoids, and anti–interleukin-1
and anti–interleukin-6 therapies. Patients with multisystem inflammatory syndrome
very clearly meet the definition of cytokine storm, since SARS-CoV-2 is no longer
present; however, it is unclear whether the cytokine storm is a driver of Covid-19
or a secondary process. Furthermore, it is now clear that patients with SARS-CoV-2
infection can be asymptomatic or can have acute Covid-19 with heterogeneous severity,
a chronic course of Covid-19, or multisystem inflammatory syndrome. A critical question
concerns the factors that contribute to the severe cytokine storm–like phenotype observed
in a small fraction of patients. Coexisting conditions such as hypertension, diabetes,
and obesity are associated with more severe cases of Covid-19, possibly because of
the preexisting chronic inflammatory state or a lower threshold for the development
of organ dysfunction from the immune response.
Several important differences in therapeutic considerations should be noted between
Covid-19–associated cytokine storm and many other cytokine storm disorders. First,
cytokine storm triggered by infection with SARS-CoV-2 may require different therapies
from those used for cytokine storm due to other causes. Cytokines may be both a key
component of the cytokine storm and an essential factor in the antimicrobial response.
Thus, blocking cytokine signaling may actually impair clearance of SARS-CoV-2, increase
the risk of secondary infections, and lead to worse outcomes, as seen with influenza
virus.
96
Since interleukin-6 and other cytokines are potentially critical for both a healthy
response to SARS-CoV-2 and a detrimental cytokine storm, it is particularly important
that the right subgroups of patients with Covid-19 are selected for treatments at
the right time. Despite positive anecdotal reports, two large, randomized, controlled
trials of anti–interleukin-6 receptor antibody therapies did not show a survival benefit
in hospitalized patients with Covid-19.
97,98
Second, the primary site of infection and disease most likely contributes to differences
in immune responses and mechanisms underlying the cytokine storm, which have implications
for treatment. For example, selective elimination of the primary viral reservoir is
beneficial in patients with HHV-8–associated multicentric Castleman’s disease but
is not possible in patients with Covid-19.
Third, lymphopenia is not often observed in cytokine storm disorders, but it is a
hallmark of severe Covid-19. It is currently unclear whether the lymphopenia observed
in Covid-19 is due to tissue infiltration or destruction of lymphocytes.
Fourth, clotting issues can occur across cytokine storm disorders, but thromboembolic
events appear to be more frequent in Covid-19–associated cytokine storm.
99
Finally, although cytokine panels have not been measured simultaneously on the same
platform across Covid-19–associated cytokine storm and other cytokine storm disorders,
preliminary results suggest that circulating levels of several cytokines, such as
interleukin-6, as well as other inflammatory markers, such as ferritin, are less severely
elevated in Covid-19 than in some of the other cytokine storm disorders.
26
Levels of inflammatory mediators in pulmonary tissue during infection with SARS-CoV-2
remain unknown.
Despite the many unknowns, a recent randomized, controlled trial showing that dexamethasone
reduces mortality among the most severe cases of Covid-19, characterized by elevated
CRP levels and supplemental oxygen requirements, and potentially worsens outcomes
in milder cases suggests that excessive, late-stage inflammation contributes to mortality.
88
A meta-analysis of seven randomized trials showed that 28-day all-cause mortality
in critically ill patients with Covid-19 was lower among those who were treated with
glucocorticoids than among those who received usual care or placebo.
100
An observational study suggesting that patients with Covid-19 have a good response
to glucocorticoids when the CRP level is high but a poor response when the level is
low is consistent with these findings.
101
Further support comes from positive anecdotal reports of targeted antagonists against
interleukin-1, granulocyte–macrophage colony-stimulating factor, and JAK1 and JAK2
in patients with Covid-19.
102-105
Likewise, the observation that proinflammatory agents such as inhaled interferon-β
have a positive effect if given early in the disease course is consistent with a model
in which immunostimulation that enhances antiviral activity is helpful early (and
probably harmful late), whereas immunosuppression is helpful late and harmful early.
As with dexamethasone, the timing of treatment and selection of subgroups of patients
included in studies will most likely have an effect on outcomes.
Despite unknowns regarding the role of immune dysregulation and cytokine storm in
Covid-19, hundreds of immunomodulatory drugs are currently under investigation.
102
Many of these treatments have been used for other cytokine storm disorders. Canakinumab,
an anti–interleukin-1β monoclonal antibody, and anakinra are both being studied for
Covid-19–induced ARDS. Acalabrutinib, a selective inhibitor of Bruton tyrosine kinase
that regulates B-cell and macrophage signaling and activation, may have promise for
dampening the hyperinflammatory response in Covid-19.
106
JAK1 and JAK2 inhibitors, which are approved for the treatment of a number of autoimmune
and neoplastic conditions, have the potential to inhibit signaling downstream of type
I interferon, interleukin-6 (and other gp130 family receptors), interferon-γ, and
interleukin-2, among other cytokines.
107
Much like anti–interleukin-6 antibody therapy, inhibition of Bruton tyrosine kinase
and JAK could prove to be damaging or unhelpful if given too soon, when the immune
response to SARS-CoV-2 is critical in controlling viral replication and clearance.
Therapeutics
The general treatment strategy for cytokine storm involves supportive care to maintain
critical organ function, control of the underlying disease and elimination of triggers
for abnormal immune system activation, and targeted immunomodulation or nonspecific
immunosuppression to limit the collateral damage of the activated immune system. As
noted throughout this review, a number of drugs are effective across multiple disorders
under the cytokine storm umbrella and still more may be effective in multiple conditions
that have not yet been studied.
Given the growing number of new therapeutics targeting various aspects of the immune
system and our ability to probe the biologic mechanisms of disease, further research
should focus on the identification of drugs that can be used across cytokine storm
disorders and precision diagnostics for selecting the right drugs for the right patients,
regardless of the underlying condition.
108,109
A study involving patients with systemic juvenile idiopathic arthritis revealed subgroups
of patients with cytokine profiles in which interleukin-6 and interleukin-18 predominated,
pointing toward available therapeutic approaches.
110
Likewise, biomarkers were recently shown to effectively predict which patients with
adult-onset Still’s disease would have a response to anakinra or tocilizumab.
111
The progress made in precision oncology suggests that similar efforts across cytokine
storm disorders are warranted to identify specific therapeutic targets and signatures
of response to certain drugs that cross disease boundaries. JAK signaling is an interesting
target in cytokine storm, because multiple cytokine–receptor pairs can be targeted
simultaneously, an approach that may be effective for multiple diseases driven by
different cytokines. In addition, plasma exchange and plasma filtration columns for
the adsorption of cytokines are both under evaluation for cytokine storm disorders.
It is important to consider several factors in managing cytokine storm. Neutralization
of a particular cytokine whose level is elevated in the circulation with an existing
agent (anti–interleukin-6, anti-TNF, anti–interferon-γ, or anti–interleukin-1β antibody)
will not always be effective, and blocking a cytokine with a low or normal circulating
level can be effective if it is a key component of the hyperinflammatory circuit or
if its level is potentially elevated in tissue. In addition, the various therapies
mentioned in this review have distinctive side-effect and risk profiles. All targeted
agents have target-specific risks, and combination therapy has more potential risks
than single-agent therapy. Furthermore, pathologic hyperinflammation itself is an
immunodeficiency that can put patients at risk for infections, and immunosuppressive
agents most likely increase the risk further. In this age of cytokine profiling and
individualized medicine, patients must be monitored and given appropriate prophylaxis
when treated empirically, and randomized, controlled trials should always be performed
to assess efficacy and safety.
Advancing the research and treatment of cytokine storm will require pooling of samples
for “omics” studies and collaboration among experts across conditions. The introduction
of an International Classification of Diseases, 10th Revision, code for cytokine release
syndrome in 2021 should facilitate electronic health record–based research into its
natural history, pathogenesis, and treatments. Once sufficient scientific progress
has been achieved toward biomarker-guided, individualized treatment of cytokine storm,
reliable, quick, and accessible assays will be needed to measure soluble mediators
of inflammation in plasma and tissues.
Summary
Mild, secondary organ dysfunction during an inflammatory response is evolutionarily
acceptable if it allows the host to overcome the infection and survive. If the inflammatory
response causes excessive organ dysfunction that puts host survival and reproductive
fitness at risk (in the absence of ventilatory support and dialysis), then it is pathologic.
Extensive regulatory mechanisms exist that modulate the immune response and prevent
cytokine storm. Nevertheless, the disorder can still occur due to iatrogenic causes,
pathogens, cancers, autoimmunity, and autoinflammatory mechanisms. Distinguishing
between protective inflammatory responses and pathologic cytokine storm has important
implications for treatment and is quite challenging. No unifying definition of cytokine
storm exists, and there is much disagreement about what the definition should be and
whether specific conditions such as Covid-19 should be included in the spectrum of
cytokine storm disorders. We propose a unifying definition for cytokine storm that
is based on the following criteria: elevated circulating cytokine levels, acute systemic
inflammatory symptoms, and secondary organ dysfunction beyond that which could be
attributed to a normal response to a pathogen, if a pathogen is present. Targeted
therapeutic approaches to cytokine storm associated with idiopathic multicentric Castleman’s
disease, HLH, or CAR T-cell therapy have turned deadly conditions into often reversible
states. Given advances in “multi-omic” profiling and therapeutic modulation of the
immune system, as well as concerted efforts to work across the cytokine storm umbrella,
we expect to see continued improvements in outcomes.
Related collections
Most cited references114
- Record: found
- Abstract: found
- Article: not found
Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China
Chaolin Huang, Yeming Wang, Xingwang Li … (2020)
- Record: found
- Abstract: found
- Article: not found
Dexamethasone in Hospitalized Patients with Covid-19 — Preliminary Report
Mark Peters (2021)

- Record: found
- Abstract: found
- Article: found
Autoantibodies against type I IFNs in patients with life-threatening COVID-19
Paul Bastard, Lindsey B. Rosen, Qian Zhang … (2021)
Author and article information
Comments
Comment on this article
scite_
0
0
0
0
0
0
0
0
Citing PublicationsSupportingMentioningContrasting
See how this article has been cited at scite.ai
scite shows how a scientific paper has been cited by providing the context of the citation, a classification describing whether it supports, mentions, or contrasts the cited claim, and a label indicating in which section the citation was made.