- Record: found
- Abstract: found
- Article: found
Crossing the Rift valley: using complete mitogenomes to infer the diversification and biogeographic history of ethiopian highlands Ptychadena (anura: Ptychadenidae)

Read this article at
Abstract
The Ethiopian Highlands are considered a biodiversity hotspot, harboring a high number of endemic species. Some of the endemic species probably diversified in situ; this is, for example, the case of a monophyletic clade containing 12 known species of grass frogs of the genus Ptychadena. The different species occur at elevations ranging from 1,500 to above 3,400 m and constitute excellent models to study the process of diversification in the highlands as well as adaptations to high elevations. In this study, we sampled 294 specimens across the distribution of this clade and used complete mitogenomes and genome-wide SNP data to better understand how landscape features influenced the population structure and dispersal of these grass frogs across time and space. Using phylogenetic inference, population structure analyses, and biogeographic reconstructions, we found that the species complex probably first diversified on the south-east side of the Great Rift Valley. Later on, species dispersed to the north-west side, where more recent diversification occurred. We further demonstrate that Ptychadena species have dispersed across the Great Rift Valley at different times. Our analyses allowed for a more complete understanding of the contribution of geological events, biogeographic barriers and climatic changes as drivers of species diversification and adaptation in this important biogeographic region.
Related collections
Most cited references84

- Record: found
- Abstract: found
- Article: found
Trimmomatic: a flexible trimmer for Illumina sequence data
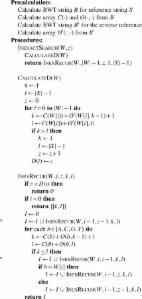
- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform

- Record: found
- Abstract: found
- Article: found