- Record: found
- Abstract: found
- Article: found
Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression
Read this article at
Abstract
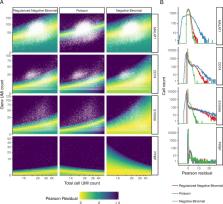
Single-cell RNA-seq (scRNA-seq) data exhibits significant cell-to-cell variation due to technical factors, including the number of molecules detected in each cell, which can confound biological heterogeneity with technical effects. To address this, we present a modeling framework for the normalization and variance stabilization of molecular count data from scRNA-seq experiments. We propose that the Pearson residuals from “regularized negative binomial regression,” where cellular sequencing depth is utilized as a covariate in a generalized linear model, successfully remove the influence of technical characteristics from downstream analyses while preserving biological heterogeneity. Importantly, we show that an unconstrained negative binomial model may overfit scRNA-seq data, and overcome this by pooling information across genes with similar abundances to obtain stable parameter estimates. Our procedure omits the need for heuristic steps including pseudocount addition or log-transformation and improves common downstream analytical tasks such as variable gene selection, dimensional reduction, and differential expression. Our approach can be applied to any UMI-based scRNA-seq dataset and is freely available as part of the R package sctransform, with a direct interface to our single-cell toolkit Seurat.
Related collections
Most cited references12

- Record: found
- Abstract: found
- Article: found
A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor

- Record: found
- Abstract: found
- Article: found
A general and flexible method for signal extraction from single-cell RNA-seq data

- Record: found
- Abstract: found
- Article: found