- Record: found
- Abstract: found
- Article: found
Quantifying label enrichment from two mass isotopomers increases proteome coverage for in vivo protein turnover using heavy water metabolic labeling
Read this article at
Abstract
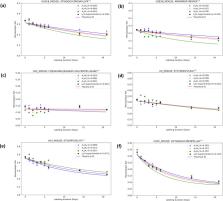
Heavy water metabolic labeling followed by liquid chromatography coupled with mass spectrometry is a powerful high throughput technique for measuring the turnover rates of individual proteins in vivo. The turnover rate is obtained from the exponential decay modeling of the depletion of the monoisotopic relative isotope abundance. We provide theoretical formulas for the time course dynamics of six mass isotopomers and use the formulas to introduce a method that utilizes partial isotope profiles, only two mass isotopomers, to compute protein turnover rate. The use of partial isotope profiles alleviates the interferences from co-eluting contaminants in complex proteome mixtures and improves the accuracy of the estimation of label enrichment. In five different datasets, the technique consistently doubles the number of peptides with high goodness-of-fit characteristics of the turnover rate model. We also introduce a software tool, d2ome+, which automates the protein turnover estimation from partial isotope profiles.
Abstract
Heavy water metabolic labeling followed by liquid chromatography coupled mass spectrometry (LC-MS) is a powerful approach to characterize in vivo protein turnover rates, however, peptide co-elution causes overlap of their isotope profiles in LC-MS and affects the proteome coverage. Here, the authors develop an approach to increase the proteome coverage for in vivo protein turnover by using partial isotope profiles from two mass isotopomers.
Related collections
Most cited references55

- Record: found
- Abstract: found
- Article: found
The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible
- Record: found
- Abstract: found
- Article: found
The Gene Ontology resource: enriching a GOld mine
- Record: found
- Abstract: found
- Article: not found