- Record: found
- Abstract: found
- Article: found
Comparative Genomics of Degradative Novosphingobium Strains With Special Reference to Microcystin-Degrading Novosphingobium sp. THN1
Read this article at
Abstract
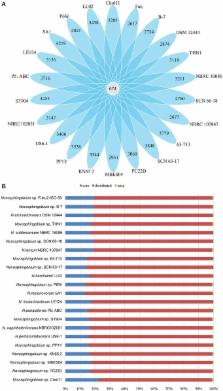
Bacteria in genus Novosphingobium associated with biodegradation of substrates are prevalent in environments such as lakes, soil, sea, wood and sediments. To better understand the characteristics linked to their wide distribution and metabolic versatility, we report the whole genome sequence of Novosphingobium sp. THN1, a microcystin-degrading strain previously isolated by Jiang et al. ( 2011) from cyanobacteria-blooming water samples from Lake Taihu, China. We performed a genomic comparison analysis of Novosphingobium sp. THN1 with 21 other degradative Novosphingobium strains downloaded from GenBank. Phylogenetic trees were constructed using 16S rRNA genes, core genes, protein-coding sequences, and average nucleotide identity of whole genomes. Orthologous protein analysis showed that the 22 genomes contained 674 core genes and each strain contained a high proportion of distributed genes that are shared by a subset of strains. Inspection of their genomic plasticity revealed a high number of insertion sequence elements and genomic islands that were distributed on both chromosomes and plasmids. We also compared the predicted functional profiles of the Novosphingobium protein-coding genes. The flexible genes and all protein-coding genes produced the same heatmap clusters. The COG annotations were used to generate a dendrogram correlated with the compounds degraded. Furthermore, the metabolic profiles predicted from KEGG pathways showed that the majority of genes involved in central carbon metabolism, nitrogen, phosphate, sulfate metabolism, energy metabolism and cell mobility (above 62.5%) are located on chromosomes. Whereas, a great many of genes involved in degradation pathways (21–50%) are located on plasmids. The abundance and distribution of aromatics-degradative mono- and dioxygenases varied among 22 Novosphingoibum strains. Comparative analysis of the microcystin-degrading mlr gene cluster provided evidence for horizontal acquisition of this cluster. The Novosphingobium sp. THN1 genome sequence contained all the functional genes crucial for microcystin degradation and the mlr gene cluster shared high sequence similarity (≥85%) with the sequences of other microcystin-degrading genera isolated from cyanobacteria-blooming water. Our results indicate that Novosphingobium species have high genomic and functional plasticity, rearranging their genomes according to environment variations and shaping their metabolic profiles by the substrates they are exposed to, to better adapt to their environments.
Related collections
Most cited references83
- Record: found
- Abstract: found
- Article: not found
ISfinder: the reference centre for bacterial insertion sequences
- Record: found
- Abstract: found
- Article: not found
Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome".

- Record: found
- Abstract: found
- Article: found