- Record: found
- Abstract: found
- Article: found
Internalization of the Membrane Attack Complex Triggers NLRP3 Inflammasome Activation and IL-1β Secretion in Human Macrophages
Read this article at
Abstract
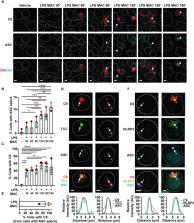
Interleukin 1β (IL-1β) plays a major role in inflammation and is secreted by immune cells, such as macrophages, upon recognition of danger signals. Its secretion is regulated by the inflammasome, the assembly of which results in caspase 1 activation leading to gasdermin D (GSDMD) pore formation and IL-1β release. During inflammation, danger signals also activate the complement cascade, resulting in the formation of the membrane attack complex (MAC). Here, we report that stimulation of LPS-primed human macrophages with sub-lytic levels of MAC results in activation of the NOD-like receptor 3 (NLRP3) inflammasome and GSDMD-mediated IL-1β release. The MAC is first internalized into endosomes and then colocalizes with inflammasome components; adapter protein apoptosis associated speck-like protein containing a CARD (ASC) and NLRP3. Pharmacological inhibitors established that MAC-triggered activation of the NLRP3 inflammasome was dependent on MAC endocytosis. Internalization of the MAC also caused dispersion of the trans-Golgi network. Thus, these data uncover a role for the MAC in activating the inflammasome and triggering IL-1β release in human macrophages.
Related collections
Most cited references63
- Record: found
- Abstract: found
- Article: not found
Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death.
- Record: found
- Abstract: found
- Article: not found