- Record: found
- Abstract: found
- Article: found
Discovery of novel quinazoline derivatives bearing semicarbazone moiety as potent EGFR kinase inhibitors
Read this article at
Abstract
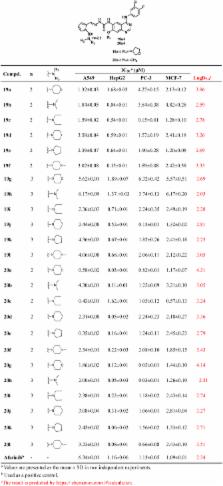
Aimed at discovering effective EGFR inhibitors, six series of quinazoline derivatives bearing a semicarbazone moiety were designed, synthesized and evaluated in different cancer cell lines (A549, HepG2, MCF-7 and PC-3). Most of the selected compounds showed remarkable cytotoxicity with IC 50 values reaching the nanomole range. Further, the inhibition efficacy of 11 compounds against EGFR kinases was tested, which demonstrated excellent IC 50 values in nanomolar level. Importantly, 2 compounds exhibited IC 50 values of 0.05 nM and 0.1 nM against wild type EGFR respectively, suggesting more potent activities than that of the positive control, Afatinib (4.0 nM). Excitingly, 2 compounds showed excellent enzyme inhibitory activity with 8.6 nM and 5.6 nM for double T790 M/L858R mutant EGFRs, which is almost the same as Afatinib (3.8 nM). Structure–activity relationships (SARs) analysis indicated that the type of small molecule amine in pyrrole moiety or the chain length of pyrrolamine moiety had no obvious impact on the inhibition efficacy of our synthesized compounds against cancer cells. In addition, results of cell cycle analysis indicated that the G2/M phase of A549 cells was efficiently arrested by the selected compounds. These preliminary results demonstrate that 2 compounds may be promising lead compound-targeting EGFR.
Graphical abstract

Highlights
-
•
Six series of quinazoline derivatives bearing semicarbazone moiety were designed and synthesized.
-
•
Most of the selected compounds showed excellent cytotoxicity activity with the IC50 values.
-
•
Compound 2 exhibits excellent kinase inhibitory activity against EGFR WT and EGFR T790M/L858R.
-
•
AO staining, cell cycle analysis and docking study were carried out.
Related collections
Most cited references17
- Record: found
- Abstract: found
- Article: not found
FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets.
- Record: found
- Abstract: found
- Article: not found