- Record: found
- Abstract: found
- Article: found
Adaptive RAxML-NG: Accelerating Phylogenetic Inference under Maximum Likelihood using Dataset Difficulty

Read this article at
Abstract
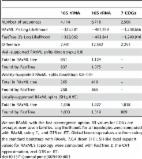
Phylogenetic inferences under the maximum likelihood criterion deploy heuristic tree search strategies to explore the vast search space. Depending on the input dataset, searches from different starting trees might all converge to a single tree topology. Often, though, distinct searches infer multiple topologies with large log-likelihood score differences or yield topologically highly distinct, yet almost equally likely, trees. Recently, Haag et al. introduced an approach to quantify, and implemented machine learning methods to predict, the dataset difficulty with respect to phylogenetic inference. Easy multiple sequence alignments (MSAs) exhibit a single likelihood peak on their likelihood surface, associated with a single tree topology to which most, if not all, independent searches rapidly converge. As difficulty increases, multiple locally optimal likelihood peaks emerge, yet from highly distinct topologies. To make use of this information, we introduce and implement an adaptive tree search heuristic in RAxML-NG, which modifies the thoroughness of the tree search strategy as a function of the predicted difficulty. Our adaptive strategy is based upon three observations. First, on easy datasets, searches converge rapidly and can hence be terminated at an earlier stage. Second, overanalyzing difficult datasets is hopeless, and thus it suffices to quickly infer only one of the numerous almost equally likely topologies to reduce overall execution time. Third, more extensive searches are justified and required on datasets with intermediate difficulty. While the likelihood surface exhibits multiple locally optimal peaks in this case, a small proportion of them is significantly better. Our experimental results for the adaptive heuristic on 9,515 empirical and 5,000 simulated datasets with varying difficulty exhibit substantial speedups, especially on easy and difficult datasets ( of total MSAs), where we observe average speedups of more than . Further, approximately of the inferred trees using the adaptive strategy are statistically indistinguishable from the trees inferred under the standard strategy (RAxML-NG).
Related collections
Most cited references26
- Record: found
- Abstract: found
- Article: found
RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies

- Record: found
- Abstract: found
- Article: found
IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies
- Record: found
- Abstract: found
- Article: found