- Record: found
- Abstract: found
- Article: found
Revealing the gut bacteriome of Dendroctonus bark beetles (Curculionidae: Scolytinae): diversity, core members and co-evolutionary patterns
Read this article at
Abstract
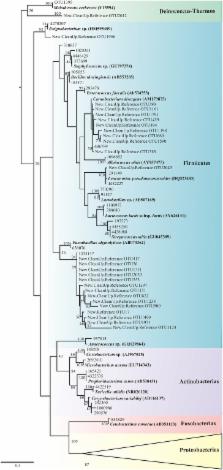
Dendroctonus bark beetles comprise 20 taxonomically recognized species, which are one of the most destructive pine forest pests in North and Central America, and Eurasia. The aims of this study were to characterize the gut bacterial diversity, to determine the core bacteriome and to explore the ecological association between these bacteria and bark beetles. A total of five bacterial phyla were identified in the gut of 13 Dendroctonus species; Proteobacteria was the most abundant, followed by Firmicutes, Fusobacteria, Actinobacteria and Deinococcus-Thermus. The α-diversity was low as demonstrated in previous studies and significant differences in β-diversity were observed. The core bacteriome was composed of Enterobacter, Pantoea, Pseudomonas, Rahnella, Raoultella, and Serratia. The tanglegram between bacteria and bark beetles suggests that members of bacterial community are acquired from the environment, possibly from the host tree. These findings improve the knowledge about the bacterial community composition, and provide the bases to study the metabolic functions of these bacteria, as well as their interaction with these bark beetles.
Related collections
Most cited references45
- Record: found
- Abstract: found
- Article: not found
Rapid denoising of pyrosequencing amplicon data: exploiting the rank-abundance distribution

- Record: found
- Abstract: found
- Article: found
Dynamic Gut Microbiome across Life History of the Malaria Mosquito Anopheles gambiae in Kenya
- Record: found
- Abstract: found
- Article: not found