- Record: found
- Abstract: found
- Article: found
Material discovery by combining stochastic surface walking global optimization with a neural network†
Read this article at
Abstract
Abstract
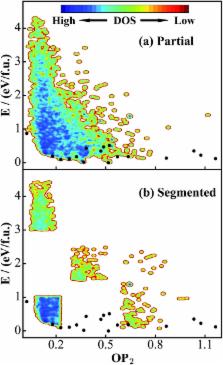
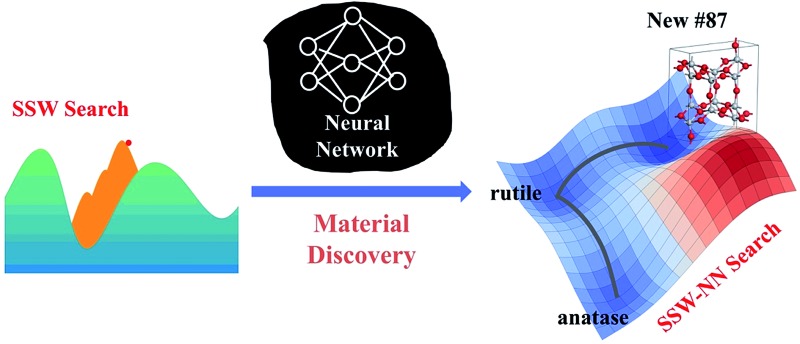
While the underlying potential energy surface (PES) determines the structure and other properties of a material, it has been frustrating to predict new materials from theory even with the advent of supercomputing facilities. The accuracy of the PES and the efficiency of PES sampling are two major bottlenecks, not least because of the great complexity of the material PES. This work introduces a “Global-to-Global” approach for material discovery by combining for the first time a global optimization method with neural network (NN) techniques. The novel global optimization method, named the stochastic surface walking (SSW) method, is carried out massively in parallel for generating a global training data set, the fitting of which by the atom-centered NN produces a multi-dimensional global PES; the subsequent SSW exploration of large systems with the analytical NN PES can provide key information on the thermodynamics and kinetics stability of unknown phases identified from global PESs. We describe in detail the current implementation of the SSW-NN method with particular focuses on the size of the global data set and the simultaneous energy/force/stress NN training procedure. An important functional material, TiO 2, is utilized as an example to demonstrate the automated global data set generation, the improved NN training procedure and the application in material discovery. Two new TiO 2 porous crystal structures are identified, which have similar thermodynamics stability to the common TiO 2 rutile phase and the kinetics stability for one of them is further proved from SSW pathway sampling. As a general tool for material simulation, the SSW-NN method provides an efficient and predictive platform for large-scale computational material screening.
Related collections
Most cited references55
- Record: found
- Abstract: found
- Article: not found
Observation of an all-boron fullerene.
- Record: found
- Abstract: found
- Article: not found
Representing potential energy surfaces by high-dimensional neural network potentials.
- Record: found
- Abstract: found
- Article: not found
Conformationally Strained trans-Cyclooctene with Improved Stability and Excellent Reactivity in Tetrazine Ligation.
Author and article information
Notes
†Electronic supplementary information (ESI) available: Derivation for the gradient of J σ with respect to NN parameters. DFT calculation setups. Parameters of atom-centered symmetry functions for generating TiO 2 NN potential. Comparison of the performance of symmetry functions with different numbers of cutoff radii. Comparison of the computational cost of NN and DFT calculations. Comparison of structural properties of experimentally known TiO 2 phase using NN and DFT PES. XYZ coordination of structures. See DOI: 10.1039/c7sc01459g