- Record: found
- Abstract: found
- Article: found
A cluster of multidrug-resistant Mycobacterium tuberculosis among patients arriving in Europe from the Horn of Africa: a molecular epidemiological study
Read this article at
Summary
Background
The risk of tuberculosis outbreaks among people fleeing hardship for refuge in Europe is heightened. We describe the cross-border European response to an outbreak of multidrug-resistant tuberculosis among patients from the Horn of Africa and Sudan.
Methods
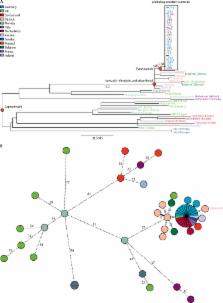
On April 29 and May 30, 2016, the Swiss and German National Mycobacterial Reference Laboratories independently triggered an outbreak investigation after four patients were diagnosed with multidrug-resistant tuberculosis. In this molecular epidemiological study, we prospectively defined outbreak cases with 24-locus mycobacterial interspersed repetitive unit-variable number tandem repeat (MIRU-VNTR) profiles; phenotypic resistance to isoniazid, rifampicin, ethambutol, pyrazinamide, and capreomycin; and corresponding drug resistance mutations. We whole-genome sequenced all Mycobacterium tuberculosis isolates and clustered them using a threshold of five single nucleotide polymorphisms (SNPs). We collated epidemiological data from host countries from the European Centre for Disease Prevention and Control.
Findings
Between Feb 12, 2016, and April 19, 2017, 29 patients were diagnosed with multidrug-resistant tuberculosis in seven European countries. All originated from the Horn of Africa or Sudan, with all isolates two SNPs or fewer apart. 22 (76%) patients reported their travel routes, with clear spatiotemporal overlap between routes. We identified a further 29 MIRU-VNTR-linked cases from the Horn of Africa that predated the outbreak, but all were more than five SNPs from the outbreak. However all 58 isolates shared a capreomycin resistance-associated tlyA mutation.
Interpretation
Our data suggest that source cases are linked to an M tuberculosis clone circulating in northern Somalia or Djibouti and that transmission probably occurred en route before arrival in Europe. We hypothesise that the shared mutation of tlyA is a drug resistance mutation and phylogenetic marker, the first of its kind in M tuberculosis sensu stricto.
Funding
The Swiss Federal Office of Public Health, the University of Zurich, the Wellcome Trust, National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC), the Medical Research Council, BELTA-TBnet, the European Union, the German Center for Infection Research, and Leibniz Science Campus Evolutionary Medicine of the Lung (EvoLUNG).
Related collections
Most cited references12

- Record: found
- Abstract: found
- Article: found
Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data
- Record: found
- Abstract: found
- Article: not found
Mutation of tlyA confers capreomycin resistance in Mycobacterium tuberculosis.
- Record: found
- Abstract: found
- Article: not found