- Record: found
- Abstract: found
- Article: found
Characterization of Differentially Expressed miRNAs and Their Predicted Target Transcripts during Smoltification and Adaptation to Seawater in Head Kidney of Atlantic Salmon
Read this article at
Abstract
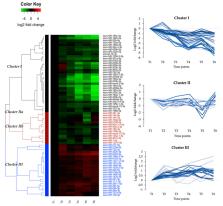
Smoltification and early seawater phase are critical developmental periods with physiological and biochemical changes in Atlantic salmon that facilitates survival in saltwater. MicroRNAs (miRNAs) are known to have important roles in development, but whether any miRNAs are involved in regulation of gene expression during smoltification and the adaption to seawater is largely unknown. Here, small RNA sequencing of materials from head kidney before, during smoltification and post seawater transfer were used to study expression dynamics of miRNAs, while microarray analysis was applied to study mRNA expression dynamics. Comparing all timepoints, 71 miRNAs and 2709 mRNAs were identified as differentially expressed (DE). Hierarchical clustering analysis of the DE miRNAs showed three major clusters with characteristic expression changes. Eighty-one DE mRNAs revealed negatively correlated expression patterns to DE miRNAs in Cluster I and III. Furthermore, 42 of these mRNAs were predicted as DE miRNA targets. Gene enrichment analysis of negatively correlated target genes showed they were enriched in gene ontology groups hormone biosynthesis, stress management, immune response, and ion transport. The results strongly indicate that post-transcriptional regulation of gene expression by miRNAs is important in smoltification and sea water adaption, and this study identifies several putative miRNA-target pairs for further functional studies.
Related collections
Most cited references69
- Record: found
- Abstract: found
- Article: not found
STAR: ultrafast universal RNA-seq aligner.
- Record: found
- Abstract: not found
- Article: not found
Cutadapt removes adapter sequences from high-throughput sequencing reads
- Record: found
- Abstract: found
- Article: not found