- Record: found
- Abstract: found
- Article: found
Cryptic diversity of a widespread global pathogen reveals expanded threats to amphibian conservation
Read this article at
Significance
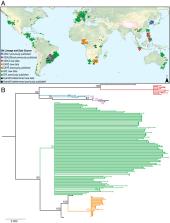
Batrachochytrium dendrobatidis [ Bd] is one of the most devastating wildlife pathogens ever documented. Most surveys for Bd report only the presence/absence of the pathogen. However, Bd has distinct genetic lineages that vary in geographic extent and virulence, thus reporting Bd presence alone is not particularly informative. Our study uses a custom method for genotyping degraded Bd DNA samples, such as those nondestructively collected from live animal or museum specimen skin swabs, and presents the discovery of a divergent lineage of Bd— BdASIA3. This study advances our understanding of the evolutionary origins of Bd, highlights areas of the world where Bd lineages are coming into contact, and opens the door to affordable, rapid genetic monitoring of this pathogen.
Abstract
Biodiversity loss is one major outcome of human-mediated ecosystem disturbance. One way that humans have triggered wildlife declines is by transporting disease-causing agents to remote areas of the world. Amphibians have been hit particularly hard by disease due in part to a globally distributed pathogenic chytrid fungus ( Batrachochytrium dendrobatidis [ Bd]). Prior research has revealed important insights into the biology and distribution of Bd; however, there are still many outstanding questions in this system. Although we know that there are multiple divergent lineages of Bd that differ in pathogenicity, we know little about how these lineages are distributed around the world and where lineages may be coming into contact. Here, we implement a custom genotyping method for a global set of Bd samples. This method is optimized to amplify and sequence degraded DNA from noninvasive skin swab samples. We describe a divergent lineage of Bd, which we call BdASIA3, that appears to be widespread in Southeast Asia. This lineage co-occurs with the global panzootic lineage ( BdGPL) in multiple localities. Additionally, we shed light on the global distribution of BdGPL and highlight the expanded range of another lineage, BdCAPE. Finally, we argue that more monitoring needs to take place where Bd lineages are coming into contact and where we know little about Bd lineage diversity. Monitoring need not use expensive or difficult field techniques but can use archived swab samples to further explore the history—and predict the future impacts—of this devastating pathogen.
Related collections
Most cited references28
- Record: found
- Abstract: not found
- Article: not found
Emerging Infectious Diseases of Wildlife-- Threats to Biodiversity and Human Health
- Record: found
- Abstract: found
- Article: not found
Colloquium paper: are we in the midst of the sixth mass extinction? A view from the world of amphibians.
- Record: found
- Abstract: not found
- Article: not found