- Record: found
- Abstract: found
- Article: found
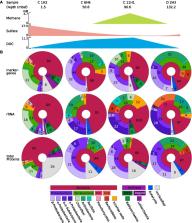
First shotgun metagenomics study of Juan de Fuca deep-sea sediments reveals distinct microbial communities above, within, between, and below sulfate methane transition zones
Read this article at
Abstract
The marine deep subsurface is home to a vast microbial ecosystem, affecting biogeochemical cycles on a global scale. One of the better-studied deep biospheres is the Juan de Fuca (JdF) Ridge, where hydrothermal fluid introduces oxidants into the sediment from below, resulting in two sulfate methane transition zones (SMTZs). In this study, we present the first shotgun metagenomics study of unamplified DNA from sediment samples from different depths in this stratified environment. Bioinformatic analyses showed a shift from a heterotrophic, Chloroflexota-dominated community above the upper SMTZ to a chemolithoautotrophic Proteobacteria-dominated community below the secondary SMTZ. The reintroduction of sulfate likely enables respiration and boosts active cells that oxidize acetate, iron, and complex carbohydrates to degrade dead biomass in this low-abundance, low-diversity environment. In addition, analyses showed many proteins of unknown function as well as novel metagenome-assembled genomes (MAGs). The study provides new insights into microbial communities in this habitat, enabled by an improved DNA extraction protocol that allows a less biased view of taxonomic composition and metabolic activities, as well as uncovering novel taxa. Our approach presents the first successful attempt at unamplified shotgun sequencing samples from beyond 50 meters below the seafloor and opens new ways for capturing the true diversity and functional potential of deep-sea sediments.
Related collections
Most cited references111

- Record: found
- Abstract: found
- Article: found
Trimmomatic: a flexible trimmer for Illumina sequence data

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools
- Record: found
- Abstract: found
- Article: found