- Record: found
- Abstract: found
- Article: found
SMILES-based deep generative scaffold decorator for de-novo drug design
Read this article at
Abstract
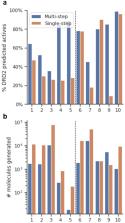
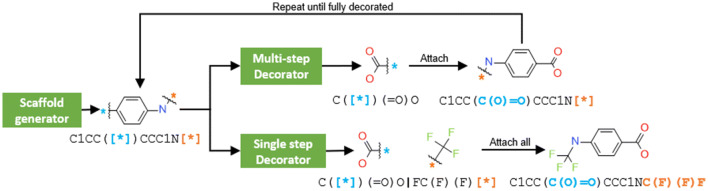
Molecular generative models trained with small sets of molecules represented as SMILES strings can generate large regions of the chemical space. Unfortunately, due to the sequential nature of SMILES strings, these models are not able to generate molecules given a scaffold (i.e., partially-built molecules with explicit attachment points). Herein we report a new SMILES-based molecular generative architecture that generates molecules from scaffolds and can be trained from any arbitrary molecular set. This approach is possible thanks to a new molecular set pre-processing algorithm that exhaustively slices all possible combinations of acyclic bonds of every molecule, combinatorically obtaining a large number of scaffolds with their respective decorations. Moreover, it serves as a data augmentation technique and can be readily coupled with randomized SMILES to obtain even better results with small sets. Two examples showcasing the potential of the architecture in medicinal and synthetic chemistry are described: First, models were trained with a training set obtained from a small set of Dopamine Receptor D2 (DRD2) active modulators and were able to meaningfully decorate a wide range of scaffolds and obtain molecular series predicted active on DRD2. Second, a larger set of drug-like molecules from ChEMBL was selectively sliced using synthetic chemistry constraints (RECAP rules). In this case, the resulting scaffolds with decorations were filtered only to allow those that included fragment-like decorations. This filtering process allowed models trained with this dataset to selectively decorate diverse scaffolds with fragments that were generally predicted to be synthesizable and attachable to the scaffold using known synthetic approaches. In both cases, the models were already able to decorate molecules using specific knowledge without the need to add it with other techniques, such as reinforcement learning. We envision that this architecture will become a useful addition to the already existent architectures for de novo molecular generation.
Related collections
Most cited references30
- Record: found
- Abstract: found
- Article: not found
The properties of known drugs. 1. Molecular frameworks.

- Record: found
- Abstract: found
- Article: found
Molecular de-novo design through deep reinforcement learning
- Record: found
- Abstract: not found
- Article: not found